临床研究服務(wù)
临床前研究服務(wù)
取得临床批件
取得生产批件或中药保护
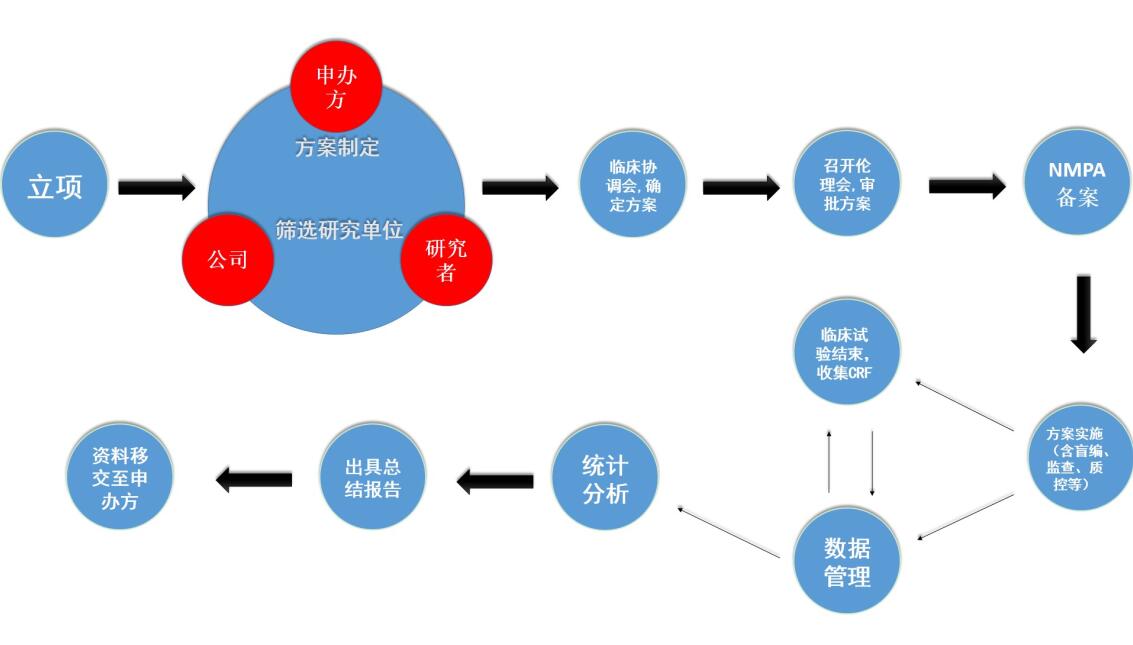
临床研究服務(wù)的主要流程如下:
(1)立项
项目成功与申办方签订合作协议后,由商(shāng)務(wù)人员对公司临床部、医學(xué)部、数据管理(lǐ)统计分(fēn)析同时进行立项,并组织召开会议,明确项目范围和申办方的需求。
(2)制定方案初稿及筛选研究单位
公司临床部负责选择临床研究单位(对每个研究单位进行详细的调研和评估),其中一家研究单位作為(wèi)组長(cháng)单位(个别申办方会建议某家研究单位作為(wèi)组長(cháng)单位),同时联系确定参加单位。医學(xué)部负责研究方案初稿的拟定,统计人员进行方案中有(yǒu)关统计學(xué)内容的撰写。
(3)召开临床研究协调会
公司、申办者与所有(yǒu)研究单位一起召开临床研究协调会,讨论并确定临床研究方案。
(4)伦理(lǐ)委员会审批,备案
确定临床研究方案后,报组長(cháng)单位伦理(lǐ)委员会审批。组長(cháng)单位召开伦理(lǐ)委员会审批临床研究方案等资料,取得组長(cháng)单位的伦理(lǐ)委员会批件后,将研究方案等资料提交申办者向國(guó)家药监局及相关省药监局进行备案登记。如果组長(cháng)单位伦理(lǐ)委员会审核时对方案等资料提出修改意见的,则需在方案等资料修改后再次召开伦理(lǐ)委员会审批同意才能(néng)实施。除组長(cháng)单位外,其他(tā)参加单位会视情况召开分(fēn)中心伦理(lǐ)审查会议对研究方案等资料进行审批。如果参加单位对临床试验方案等资料提出异议,则需反馈至组長(cháng)单位及申办方再次对方案进行讨论和修改。如果参加单位对最终确定的临床研究方案等资料还有(yǒu)异议,则可(kě)以选择退出该项目的临床研究。临床方案确定后,还需要将组長(cháng)单位的伦理(lǐ)委员会批件、临床研究方案、药检报告等研究资料在所有(yǒu)参加单位备案。
在获得伦理(lǐ)委员会批件后,由非参与统计分(fēn)析的统计专业工作人员对申办方提供的临床试验用(yòng)药物(wù)进行随机编盲工作。
涉及外资背景的申办方,需要在伦理(lǐ)获得批准后,申请遗传办备案,获批准后才能(néng)开展试验。
(5)临床试验开始、进行、结束
临床研究开始前,临床部与所有(yǒu)临床研究单位签订临床研究协议。临床研究协议签署后,通知申办方将临床试验用(yòng)药物(wù)送达相应研究单位。负责该项目的项目经理(lǐ)和监查员对相关研究者进行临床研究方案的培训,试验正式开始。在临床研究期间,监查员严格按照相关规定,检查入组病例是否符合方案规定的入选要求、排除标准,研究资料是否准确、及时、真实的填写,检查核对实验室数据并出具监查报告。病人出组后,监查员检查核对所有(yǒu)研究中心的病例资料,并回收研究资料和剩余临床试验用(yòng)药物(wù),清点剩余临床试验用(yòng)药物(wù)返还申办方。
(6)数据管理(lǐ)和统计分(fēn)析
回收临床研究资料后,数据管理(lǐ)员制定数据管理(lǐ)计划,根据临床试验方案编写数据核查计划,对数据进行检查,出具疑问表提交至研究者进行答(dá)疑,根据研究者答(dá)疑表对数据库进行修正。答(dá)疑结束后,召开数据审核会,会后锁定数据库(盲法试验时需进行揭盲)。统计人员根据锁定的数据库,按照统计分(fēn)析计划进行统计分(fēn)析,出具统计分(fēn)析报告。
(7)总结会、总结报告
统计分(fēn)析报告完成后,公司各业務(wù)部门(包括医學(xué)部、临床部、统计分(fēn)析人员等)与研究单位召开临床研究总结会,对统计分(fēn)析报告进行讨论和定稿,并对临床研究进行总结。由医學(xué)部根据统计分(fēn)析报告拟定总结报告及各分(fēn)中心的小(xiǎo)结报告初稿,并再次送交各研究单位进行审核定稿、签字确认,将研究者签字确认的总结报告提交各药物(wù)临床试验机构签章。临床研究总结报告、小(xiǎo)结报告等研究资料是申办者申请新(xīn)药证书和药品注册批件的重要资料。
(8)质控,资料移交
项目全过程均受质量管理(lǐ)与控制,项目开展前期在质量控制部支持下明确项目的质量管理(lǐ)要求,过程中安排定期对试验文(wén)件进行系统检查;申报资料在提交有(yǒu)关部门前,将经过质量控制部安排全面核定以保证其质量。试验完成后,最终通过质控的资料将被统一移交至申办者并签署交接清单。