









本期《袁来如此》為(wèi)系列文(wén)章《大分(fēn)子生物(wù)分(fēn)析概论》的第八篇,旨在根据已发表的文(wén)献资料,初步介绍LBA中的质量控制样品(QC,quality control)制备、认证(qualification)和批次一致性维护的相关概念和实践。
由于内容篇幅较長(cháng),本文(wén)将采取上下篇形式进行推送,敬请垂注!《袁来如此》专栏系广州博济医药微信公众号打造的科(kē)普學(xué)术专栏,内容均為(wèi)博济医药子公司深圳博瑞副总经理(lǐ)袁智博士原创。
配體(tǐ)结合测试方法(LBA)的性能(néng),在研究前方法验证和研究中样本分(fēn)析期间,體(tǐ)现在QC的效能(néng)。需要采用(yòng)严格和既定的方法合理(lǐ)地管理(lǐ)QC样品的生命周期,内容包括制备、质量控制以及QC性能(néng)的监控。
虽然在监管机构的指南和有(yǒu)关LBA的文(wén)献中有(yǒu)关于QC样品的制备和认证接受标准(acceptance criteria)的相关讨论,但此类文(wén)献只是较為(wèi)初步地讨论了这些参数,且只涉及方法开发和研究前验证的阶段,并没有(yǒu)对QC生命周期管理(lǐ)的问题和对这一过程至关重要的因素做出讨论和解答(dá)。关于新(xīn)QC批次的生产和认证(用(yòng)来保持QC批次间的一致性和防止测试结果偏移)的许多(duō)问题仍然没有(yǒu)得到解决。此外,大多(duō)数生物(wù)分(fēn)析实验室缺乏健全的方法或统计工具来监测QC趋势,而这种监测对于QC性能(néng)以及生命周期的管理(lǐ)是至关重要的。
本文(wén)旨在填补这一方面的空白,為(wèi)QC样品生命周期的管理(lǐ)及其组成部分(fēn)提供指导、建议和最佳实践。这其中包括:QC的制备、认证和其性能(néng)趋势的监控。帮助生物(wù)分(fēn)析实验室,在其标准操作程序 (SOPs)中,定义QC的认证,并制定批次间一致性的指导原则。应当在已验证的分(fēn)析方法或每个实验室的样本分(fēn)析计划中,对研究和分(fēn)析方法特异性的具體(tǐ)要求(assay- and study-specific requirements)和规范加以考虑和做出详细说明。本文(wén)主要侧重于用(yòng)于LBA定量和定性分(fēn)析中的QC,如药代动力學(xué)(PK)和抗药物(wù)抗體(tǐ)(ADA)的测试。其他(tā)类别的测试方法不在讨论的范畴。
标准品(Reference standard)、混合样本基质、QC的组成成分(fēn)、设备和生产策略都是制备用(yòng)于LBA的QC样品的关键因素。以下各节将根据这些因素展开讨论。
QC样品应使用(yòng)与研究样本基质相似,且尽可(kě)能(néng)与其接近的基质来制备。例如,如果研究样本是未过滤、未离心或未经活性炭处理(lǐ)的血清,则QC基质也应是未过滤、未离心或未经活性炭处理(lǐ)的同一物(wù)种的血清。应使用(yòng)未稀释(100%)的基质制备QC样品,以便对QC样品与研究样本采取相同的稀释步骤[如最低稀释度(minimum required dilution,MRD)]。
但不排除特殊情况,例如用(yòng)替代基质代替稀有(yǒu)的研究基质(通常很(hěn)难获得足够数量的稀有(yǒu)基质包括:脑脊液、关节腔滑液或来自某些物(wù)种的眼部基质(ocular matrices)等),但这需要适当的理(lǐ)由,并且只允许在需要保留基质(matrix conservation)的情况下使用(yòng)。缓解基质體(tǐ)量问题的推荐方法是在替代基质中准备3个QC水平中的2个,而在研究基质中制备一个浓度的QC。如果使用(yòng)替代基质,则需要证明其与研究基质具有(yǒu)等效性。最好使用(yòng)与制备标准品(calibrators)相同批次的基质来制备QC样品,以增强方法的一致性和重现性。
中间原液(intermediate stock)是用(yòng)于制备QC样品的待测物(wù)溶液,可(kě)以在下列稀释剂中制备,如水、缓冲液或有(yǒu)机介质(organic media)。当中间原液是水或缓冲液时,QC的最终成分(fēn)至少為(wèi)95%(v/v)的样本基质;当使用(yòng)有(yǒu)机溶剂(如DMSO或乙酸)制备时,最终成分(fēn)至少為(wèi)99%(v/v)的样本基质。
应当分(fēn)别制备QC样品与标准品,以防止加入待测物(wù)时的系统性误差。建议使用(yòng)不同的中间原液和不同的稀释步骤来制备QC样品和标准品,还建议在每个浓度中独立加入待测物(wù),而不是通过序列稀释(serial dilutions)高浓度的QC样品。这一点尤為(wèi)重要,因為(wèi)标准品如果是通过高浓度样品的序列稀释制备的,序列稀释QC样品和标准品能(néng)够掩盖稀释線(xiàn)性度的问题,应该避免这种操作。
在QC制备时,PK定量分(fēn)析中的标准品(reference standard)或ADA检测中的阳性对照品(positive antibody control)必须在其有(yǒu)效期内。应该单独建立QC样品的稳定性和有(yǒu)效期(与用(yòng)于制备的标准品无关),因為(wèi)QC样品中的待测物(wù)是在基质之中,与原液中的标准品是不同的。
可(kě)以制备、分(fēn)装和储存将标准品用(yòng)研究基质或适当稀释剂稀释时所产生的中间原液,以供未来制备QC样品或标准品。在这种情况下,中间原液的稳定性应覆盖其储存窗口期,即从其制备日期起至使用(yòng)日期為(wèi)止。这可(kě)以通过比较下述2种样品来实现:1. 从冻存的中间原液(frozen intermediate stock)制备的标准品和/或QC样品;2. 使用(yòng)初始的标准品原液(original reference standard stock)制备的标准品和/或QC样品。
正确地选择用(yòng)于制备QC样品的基质,对于保证分(fēn)析方法的质量和防止分(fēn)析结果漂移是至关重要的。这在ADA检测中尤其重要,因為(wèi)合格的基质(QMP),作為(wèi)阴性对照(NC),将直接影响微孔板特异性的切点,必须在已验证的定量分(fēn)析方法中或适当的SOP中,建立并明确规定适当的基质筛选(screening)、选择(selection)过程以及接受标准。有(yǒu)关基质质量认证(qualification)的建议总结如下。
第一个合格基质的批次认证通常是作為(wèi)分(fēn)析方法开发的一部分(fēn)进行的,并在方法验证时进行正式的认证。
& 认证足够體(tǐ)量的合格基质,足以在多(duō)个研究和多(duō)个药物(wù)开发阶段一直使用(yòng)。至少,有(yǒu)足够體(tǐ)量的合格基质可(kě)以满足研究前验证以及一项或多(duō)项生物(wù)样本分(fēn)析研究。
& 合格基质的储存条件与预期研究样本的储存条件一致(如-20℃或-80℃)。
& 制备混合基质(matrix pool)的第一步,是通过比较未添加和添加了待测物(wù)的基质样本所产生的分(fēn)析信号,来筛选单个基质样本或混合基质样本(多(duō)个、单个样本的混合物(wù))。
& 作為(wèi)背景值,考察未添加待测物(wù)的基质样本的响应值。应当将背景异常高的基质样本剔除,例如,高于PK定量下限(LLOQ)或高于ADA预估切点的样本。对于ADA分(fēn)析,背景异常低也可(kě)能(néng)有(yǒu)问题。
& 对于PK定量分(fēn)析,可(kě)根据分(fēn)析方法或实验室SOP中确定的接受标准来评估添加了待测物(wù)的基质样本。如建议相对误差(RE)的接受标准在±20%范围内;建议在LLOQ和低QC(LQC)之间的浓度水平上添加待测物(wù)到单个基质样本,并且至少在一次分(fēn)析运行中这样做。
& 对于ADA分(fēn)析,必须强调空白信号(blank signal,NC)的重要性。当没有(yǒu)其它批次的基质可(kě)比较时,建议在筛查测试(screen,Tier 1)中评估单个基质样本,并比较组内各个样本的响应值,以评估其原始响应值。应将所有(yǒu)异常高响应值或低响应值的单个基质排除在替换混合基质(replacement matrix pool)之外。在可(kě)能(néng)的情况下,还应将混合基质(matrix pool)与一组疾病状态的基质样本进行比较,以确定其合用(yòng)性(suitability)。
& 对于PK定量分(fēn)析,混合基质的背景的接受标准,可(kě)以是比预估的LLOQ低2-3倍的基质响应。ADA分(fēn)析的接受标准,可(kě)以是低于预估的切点或在特定的响应范围内。
& 使用(yòng)与认证第一批次相同的分(fēn)析方法,对替换批次的基质样本进行认证。
–确保未外加待测物(wù)的现有(yǒu)的和替换的合格基质(QMP)的响应值具有(yǒu)可(kě)比性。
–对于PK定量分(fēn)析,应当将替换基质批次与现有(yǒu)合格批次进行比较。至少在一次分(fēn)析运行中,分(fēn)析添加了待测物(wù)标准品的样本(浓度在LLOQ和LQC之间,至少n=3)。标准品(reference standard)的分(fēn)析回收率(AR),在现有(yǒu)和替代批次的基质中,都应在80至120%的范围内。同时,使用(yòng)两个基质批次制备的样本所测定的标准品浓度之差不超过10%。
–為(wèi)了认证ADA分(fēn)析中的替换合格基质(replacement QMP),应当使用(yòng)先前建立的微孔板特异的切点因子,对单个基质样本进行筛查。筛查测试中是阳性的所有(yǒu)单个样本都应排除在替换合格基质之外。另一种方法是直接考察每个基质样本的信噪比(S/N),应将超过经过验证的切点因子的基质样本排除在替换合格基质之外。
以下推荐的是替换合格基质的接受标准: 替换基质批次的响应值,应当在现有(yǒu)基质批次响应值的±10%以内。如果不是,则应当评估一组单个基质样本,并与两个微孔板特异的切点比较,以确定筛查结果是阳性或阴性。其中一个切点是用(yòng)现有(yǒu)合格基质的阴性对照计算的,另一个切点是用(yòng)替换合格基质制备的阴性对照计算的。使用(yòng)这两个切点(现有(yǒu)vs.替换微孔板特异的切点)所得出的筛查结果(阴性/阳性状态)应当相似(comparable),但borderline samples,根据基质响应,可(kě)能(néng)是阳性或阴性。
在PK分(fēn)析中,基质的背景响应应保持在最低状态,因為(wèi)它影响分(fēn)析灵敏度,并可(kě)能(néng)限制定量的范围。对于ADA分(fēn)析,建议一旦获得验证数据,就应尽快确定基质响应范围的上限和下限。
& 在非竞争性定量(如PK)免疫测试中,基质背景不应超过LLOQ的1/3。
& 在竞争性免疫分(fēn)析中,背景值至少為(wèi)最低校准点数值的1.11倍。这是基于B/B0(最低校准点信号/零校准点信号,lowest calibrator signal/zero calibrator)建议的90%(100/90)。
& 在ADA和其他(tā)非定量分(fēn)析中,对基质背景的一般建议是相对发光单位(RLUs)≤200和吸光度≤0.200。有(yǒu)些测试方法的基质背景则比上述建议的要高。
& 基质背景的下限由仪器响应决定,不同仪器和不同实验室的仪器响应可(kě)能(néng)不同。
& 用(yòng)于制备QC样品的设备,包括移液器,应进行精密度验证,并应在校准范围内。
& 设备校准的过程和频率应在SOP中规定明示。
& 在一些实验室,除了定期校准之外,还需要在使用(yòng)移液器制备QC样品之前进行额外的校准检查,但在一些实验室,如果定期校准仍然有(yǒu)效,则不需要重新(xīn)进行校准检查。
此处的一般运行接受标准适用(yòng)于现有(yǒu)和替换QC批次的认证。此处下列术语:基線(xiàn)批次(baseline QC lots)、遗留QC批次(legacy lots)或QC比较批次(comparator QC lots)可(kě)互换使用(yòng)。
1. QC样品可(kě)与之前认证过的,且有(yǒu)已知稳定性的冻存校准品进行比对和认证。如果没有(yǒu)合格的冻存标准品,则应使用(yòng)重新(xīn)制备的标准曲線(xiàn)对QC样品进行评估。在后一种方法中,需要加入之前认证过的一组QC样品对新(xīn)鲜制备的标准曲線(xiàn)进行认证。
2. 认证(qualification)是通过评估分(fēn)析内和分(fēn)析间的精密度(inter- and intra-assay precision,对所有(yǒu)方法)和准确度(对定量方法)来进行的。
3. 对定量和药代动力學(xué)分(fēn)析方法,QC样品必须满足精密度和准确度标准:变异系数(coefficient of variation,CV) ≤20%,RE在±20%之内;定量下限(LLOQ)和上限(ULOQ):CV≤25%,RE在±25%之内。
1. QC样品应满足CV≤20%的标准。
2. 现有(yǒu)和替换QC的响应值都应在其相应浓度级别已建立的响应范围内。如果替换的QC批次不在相应的信号范围内,则实验室必须排除故障并确定其根本原因。ADA阳性对照(PC)和阴性对照(NC)信号漂移的潜在原因包括:(a)待测物(wù)添加错误;(b) 混合基质选择不当;(c)信号范围设定不正确。
后续的故障排除应遵循以下顺序: 首先准备一个新(xīn)批次的QC样品,如果重复制备的QC仍然失败,则认证一个新(xīn)批次的合格基质(QMP);然后重新(xīn)制备阳性和阴性对照,如果新(xīn)制备的阳性和阴性对照仍然在其信号范围之外,则纳入研究中测试运行得出的额外数据,进而重新(xīn)评估已建立的信号范围。每个实验室应当根据足够的测试运行次数所得出的数据,适当地确定验证后信号范围,以避免将其范围设置得过窄或者不适合長(cháng)期使用(yòng)。
3. 接受标准:高阳性对照(HPC)>低阳性对照(LPC)>切点>阴性对照。
1. 建议在每次认证的测试运行中,至少测试3组现有(yǒu)和替换QC样品。在这个要求上,个别实验室可(kě)能(néng)会有(yǒu)所不同。
2. 至少三分(fēn)之二的测试运行应满足上述规定的接受标准。这些认证要求和建议见表I。
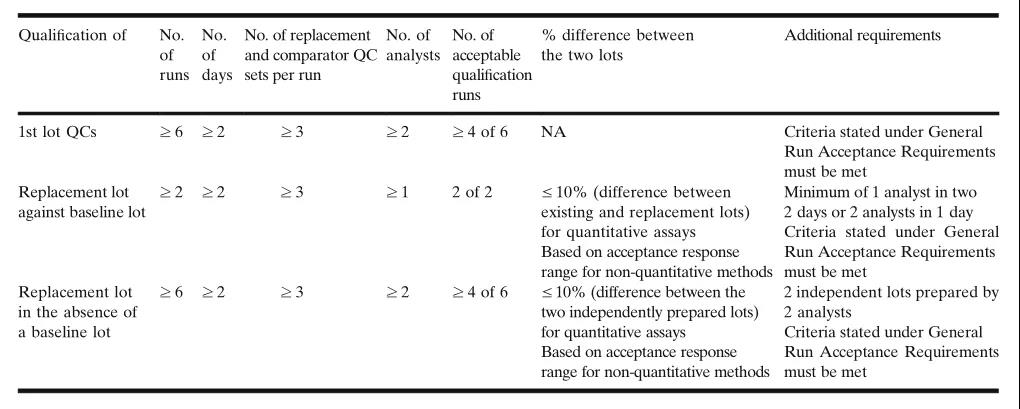
3. 现有(yǒu)的和替换的QC样品必须满足上述的常规运行接受标准。
4. 如果替换批次的QC样品不符合上述接受标准,而现有(yǒu)批次合格时,则应将替换批次废除,并制备另一批次的QC样品。当一个浓度水平的替换QC样本不合格时,可(kě)以重新(xīn)制备该浓度水平的QC。
5. 如果现有(yǒu)的QC批次在替换批次的认证过程中未能(néng)通过接受标准,则应重新(xīn)分(fēn)析以确认相关结果。如果现有(yǒu)的批次在重复分(fēn)析后仍然不合格,那么它就不能(néng)用(yòng)作比对品(comparator)。在这种情况下,应遵循<现有(yǒu)合格批次不存在时的认证>章节所描述的程序和质量标准。
第一个批次的QC样品,也被称為(wèi)基線(xiàn)(baseline)或遗留(legacy)批次,通常作為(wèi)研究前方法验证中的准确度和精密度(A&P)评估的一部分(fēn)进行认证。对研究前方法验证中的QC批次进行认证时,推荐至少进行独立运行6次,每个QC浓度至少运行3次,至少分(fēn)2天进行,至少2名分(fēn)析人员参与。且要求至少4/6的QC认证运行应该满足接受标准,才满足对该批次QC认证(见表1)。在可(kě)能(néng)的情况下,还建议将该QC批次与方法开发时所用(yòng)的QC样品建立桥接联系。对这种桥接性评价的接受标准将由各个独立的实验室自行制定, 推荐二者之间的误差為(wèi)±10%或更低。特定样品的运行接受标准必须满足常规运行接受标准。
i. 通过与现有(yǒu)合格QC批次比较的认证
在研究前的方法验证之外,每次制备替换批次QC样品时,通过与之前认证了的(合格)批次的比较,对替换批次进行认证。為(wèi)了可(kě)靠地进行可(kě)比性评估,必须使用(yòng)相同的冻存或新(xīn)鲜制备的标准曲線(xiàn)来评估现有(yǒu)的和替换的QC批次。
一个替换批次的QC样品至少要经过2个独立的认证运行(qualification runs)。QC认证运行可(kě)能(néng)在同一天由多(duō)个分(fēn)析人员(至少2名),或同一分(fēn)析人员在不同天(至少2天)进行。建议在同一认证运行中,至少要包括3组独立的替换批次QC样品和至少3组现有(yǒu)批次的QC样品。一组从单独冻存和分(fēn)装的QC所制备的样品被定义為(wèi)一组独立的QC样品(一组有(yǒu)三个浓度级别)。在QC认证过程中,不鼓励在同一测试运行中使用(yòng)同一冻存和分(fēn)装的QC制备的一组QC。个别实验室可(kě)以制定不同于此处建议的标准,如根据其对所涉及的变异性和风险的评估,分(fēn)别进行3次独立测试运行,而不是2次。
定量LBA方法中,替换QC批次的认证,应当以常规运行接受标准中的规定為(wèi)基础,同时也应当以其与现有(yǒu)合格QC批次的可(kě)比性(comparability)為(wèi)基础。例如,可(kě)比性标准可(kě)包括现有(yǒu)批次和替换批次之间±10%或以下的差异。使用(yòng)现有(yǒu)和替换QC样品的测量浓度,可(kě)以用(yòng)下面的公式确定差异百分(fēn)比值(%)。
替换批次浓度-现在批次浓度
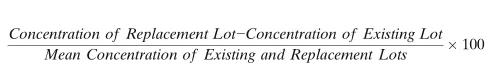
用(yòng)于ADA和其他(tā)非定量LBA检测方法, 现有(yǒu)和替换的QC批次都应该满足ADA和定性分(fēn)析的常规运行接受标准。此外,现有(yǒu)和替换PCs和NCs的响应都应该在各自建立的信号范围内。
ii. 不存在现有(yǒu)合格(认证过的)QC批次情况下的认证
可(kě)能(néng)存在这样的情况,即可(kě)能(néng)不存在遗留的,经认证合格的QC样品,因為(wèi)这些QC样品已经用(yòng)完或过期。不存在可(kě)比较QC的情况下,建议使用(yòng)不同的中间原液(intermediate stocks),由不同的分(fēn)析人员,分(fēn)别制备两个批次的QC样品。从实际操作上,可(kě)以指定一个批次,可(kě)能(néng)是批量较大的,為(wèi)主要批次(primary lot);另一批次,可(kě)能(néng)是小(xiǎo)批量的,就作為(wèi)参照品(comparator),对主要批次进行认证。对主要和次要批次的对比测试应由2人进行,每人使用(yòng)独立制备的标准曲線(xiàn)进行测试,每人进行3次测试,至少分(fēn)2天进行,总共运行6次。至少4/6 (2/3)的QC认证运行应该满足接受标准。也应满足上述的常规运行接受标准。两个替换批次的QC样品应满足±10%或更严格的偏差标准,保证二者中任何一个批次均可(kě)接受。表1总结了这些认证要求。
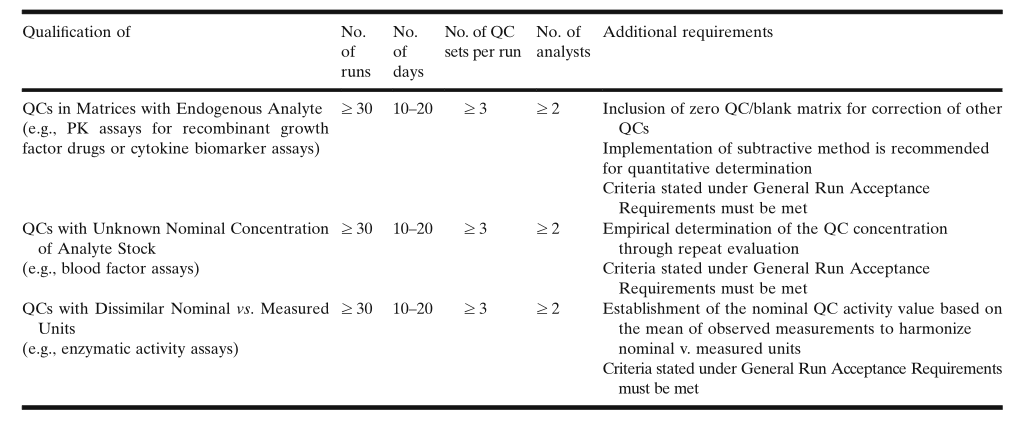
在含有(yǒu)内源性待测物(wù)的基质中制备的QC的认证
在定量PK分(fēn)析中,当基质中含有(yǒu)待测物(wù)的内源性同源物(wù)时,所测定的空白基质浓度可(kě)能(néng)是至关重要的;并应该纳入考虑。有(yǒu)2种方式:
1.减法:在报告测定的QC值之前,从测定的QC样品浓度中减去空白基质的浓度;2.加法:将测定的空白基质浓度加上QC的外加待测物(wù)浓度,以确定其调整后的标称值。Marcelletti等人介绍了几个案例研究;其中,使用(yòng)加法和减法,直接比较了加标回收率;这些案例研究评估了具有(yǒu)明显内源性水平的许多(duō)生物(wù)标记物(wù)的回收率。根据这些已发表的案例研究,减法是首选方法,而不推荐使用(yòng)加法。建议在每个认证运行中至少包含3组空白基质样本,其测定的平均浓度用(yòng)于调整值(adjusted value)的计算。建议对含有(yǒu)内源性待测物(wù)的QC样品,在10-20天内至少进行30次运行,至少有(yǒu)2人参与,每个运行使用(yòng)≥3组QC样品。此类QC认证的运行接受标准与定量PK方法的常规运行接受标准相同。
未知标称浓度(Nominal Concentration)的QC的认证
如果QC原液的标称浓度未知,如未纯化的蛋白质和一些商(shāng)业来源的对照原液(commercial control stocks),或者在血液學(xué)检测中,当crude血清/血浆被用(yòng)作血液因子的来源时,标称浓度必须根据多(duō)位分(fēn)析人员在不同日期进行的多(duō)次运行中检测到的平均值来确定。
此类测试的适当规范是特异于测试方法的,必须作為(wèi)研究前测试方法验证的一部分(fēn)建立起来。QC样品的制备方法是将待测物(wù)原液,以特定稀释倍数,加入到经过认证(合格)的空白混合基质或适当的稀释剂(diluent)中。对于所有(yǒu)成功的运行,每个QC的平均测定值就应设定為(wèi)标称值。建议至少正常实施测试运行30次,由至少2人在10-20天内执行,以设定QC样品的标称浓度。每个测试运行包含 ≥3 组QC。在最初的测定之后,日常分(fēn)析运行中QC样品的接受标准以及将来替换QC批次的接受标准為(wèi)已确定的标称值(nominal value)的±20%。任何替换批次的待测物(wù)浓度必须在此确定的范围内。由于来自血清和血浆的因子的固有(yǒu)的变异性,可(kě)能(néng)需要,基于最初的20-30次的运行数据,為(wèi)每个QC浓度水平指定一个临时的标称值;然后,在有(yǒu)其它可(kě)用(yòng)数据时,重新(xīn)评估这个标称值的适用(yòng)性。如果测试方法的内在变异性较大,则可(kě)能(néng)需要定期地重新(xīn)评估和重新(xīn)建立QC的接受范围。前述的PK定量方法的常规运行接受标准也必须得到满足。
标称浓度与测定浓度单位不一致的QC的认证
酶活性测试方法是QC标称值和测定值单位不同的方法之一。在这些分(fēn)析测试中,酶类药物(wù)的标称(外加)浓度通常是ng/mL或g/mL;然而,加标QC样品测定值的活性单位為(wèi)nmol/h/mL或nmol/h/g。这些QC单位上的差异构成了独特的挑战,因為(wèi)它不能(néng)计算准确度。
在这种情况下,可(kě)以通过建立QC的标称活性值(nominal activity value)来克服这个局限性。每个浓度级别的标称QC活性值可(kě)以,由至少2人,根据至少30次合格运行的平均测定活性值,在至少10-20天内建立。每次运行至少应包含3组QC样品。所有(yǒu)合格运行的平均活性测定值,即為(wèi)QC的标称活性值,并可(kě)以使用(yòng)下面的公式计算单个QC样品的%RE:
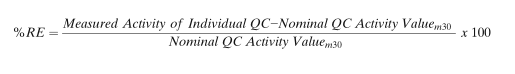
其中,标称QC活性值m30,含义是进行30次运行所测定活性的平均值。在最初的测试完成之后,日常运行和替换QC批次的接受标准就是QC活性标称值的±20%。表2总结了上面讨论的,有(yǒu)关特殊类别的QC样品的注意事项。
表2. 对特殊类别QC样品的要求的总结
特别声明
本文(wén)如有(yǒu)疏漏和误读相关指南和数据的地方,请读者评论和指正。所有(yǒu)引用(yòng)的原始信息和资料均来自已经发表學(xué)术期刊, 官方网络报道, 等公开渠道, 不涉及任何保密信息。参考文(wén)献的选择考虑到多(duō)样化但也不可(kě)能(néng)完备。欢迎读者提供有(yǒu)价值的文(wén)献及其评估。














