









本文(wén)為(wèi)大分(fēn)子生物(wù)分(fēn)析概论系列文(wén)章的第五篇,主要介绍LBA方法的平行性(parallelism)实验或研究,这是大分(fēn)子药物(wù)分(fēn)析方法所特有(yǒu)的一项研究。平行性是评估配體(tǐ)结合式测试方法(ligand-binding assay,LBA)相对准确性的关键实验。
在这个实验中,将采用(yòng)平行性研究,即通过评估稀释对生物(wù)基质中内源性待测物(wù)定量的影响,来表述一个分(fēn)析方法的相对准确性,可(kě)以评估的基本特征包括选择性、基质效应、所需的最小(xiǎo)稀释倍数、健康和患病人群的内源性待测物(wù)的水平以及LLOQ。本文(wén)将比较并讨论评估支持PK定量分(fēn)析的LBA方法中关键参数的若干方法以及每种方法的优缺点。
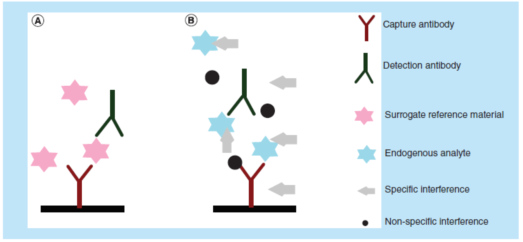
LBA定量分(fēn)析方法,是通过比较已知药物(wù)浓度的校准品与其未知浓度的生物(wù)样本的免疫反应活性(immunoreactivity)来测定生物(wù)样本中大分(fēn)子药物(wù)的浓度。对于一个性能(néng)良好的LBA方法,其校准曲線(xiàn)(合适的logistic regression拟合结果)应具有(yǒu)平行性,即能(néng)够支持如下假设:作為(wèi)测试试剂的抗體(tǐ)其结合特性足够相似、可(kě)用(yòng)于测定稀释了的样本中的待测物(wù)的浓度。导致非平行性的两个主要因素是:(1)校准参照(比)物(wù)质与未知待测物(wù)之间的免疫亲和力特征的差异(对捕获和检测试剂而言);(2)校准曲線(xiàn)基质、质量控制样品(QC)基质和研究人群的基质之间基质效应的差异(图1)。
对于旨在测定外源性蛋白药物(wù)以支持药代动力學(xué)(PK)研究的LBA方法,参照(比)标准物(wù)质通常能(néng)得到详细的表征,且具有(yǒu)全面的分(fēn)析证书(CoA)。用(yòng)作校准品的参照(比)标准物(wù)质通常与待测物(wù)相同。外加不同浓度的待测物(wù)制备的质量控制样品则用(yòng)于评估准确度、精密度、选择性、灵敏度和稀释線(xiàn)性度,以支持最终的定量分(fēn)析方法的验证。美國(guó)FDA指南草(cǎo)案、EMA指南和行业共识白皮书详细描述了进行这些实验的相关建议。目前,这些建议在用(yòng)于PK定量分(fēn)析的LBA方法验证中已得到广泛地运用(yòng)。
对于测定内源性蛋白质,如生物(wù)标志(zhì)物(wù)(biomarker)和“游离”/“总”药物(wù)靶标的LBA方法的开发和认证,相关策略将不同于PK定量分(fēn)析方法。这是因為(wèi)通常不存在纯化了的、完全表征的内源性参照(比)物(wù),且不含待测物(wù)的空白基质可(kě)能(néng)也不存在,故需要一种相对定量的方法。用(yòng)于内源性待测物(wù)如生物(wù)标志(zhì)物(wù)的LBA方法的平行性将在后续文(wén)章中作专门讨论,敬请关注。
总而言之,平行性研究是通过评估稀释对生物(wù)基质中待测物(wù)定量分(fēn)析的影响,来表述一个分(fēn)析方法相对准确性(relative accuracy)的一项关键指标。可(kě)以评估的相关分(fēn)析方法的基本特征包括选择性、基质效应、所需最低稀释倍数(minimum required dilution, MRD)、健康和患病人群中内源性待测物(wù)的浓度和LLOQ。
在过去的十多(duō)年中,科(kē)學(xué)文(wén)献中就平行性实验操作和适用(yòng)标准发表了许多(duō)讨论结果。2003年,DeSilva等人建议使用(yòng)由几个Cmax样本(已测样本再分(fēn)析样本)制成的混合样本,以支持PK方法的研究中验证。2011EMA指南建议使用(yòng)高浓度研究样本(Cmax研究样本)稀释到至少三个浓度来评估PK方法的平行性,还建议计算稀释样本之间的精密度以评估平行性。由于使用(yòng)了真实样本进行平行性研究,即研究样本中的待测物(wù)已经处于生物(wù)基质中,而不是外加待测物(wù)到空白生物(wù)基质而生成研究样本。
因此,在某种意义上用(yòng)于PK定量分(fēn)析的和用(yòng)于生物(wù)标志(zhì)物(wù)的LBA方法在平行性研究中处于相似地位。所以,虽然本文(wén)预期只讨论用(yòng)于PK定量分(fēn)析的LBA方法的平行性,但下面的讨论往往也提到或比较二者。
本文(wén)将探讨并比较相关评估关键参数的方法,包括平行性实验和其它传统的方法,讨论如何使用(yòng)平行性数据来指导分(fēn)析方法的开发以及每种策略的优缺点。
LBA定量测定存在于生物(wù)基质中的待测物(wù),无需事先萃取。在理(lǐ)想情况下,捕获/检测试剂应结合(并且仅结合)待测物(wù),保证其不会与任何其它的蛋白发生交叉反应。不幸的是,情况往往并非如此。在现实世界中,生物(wù)基质中的内源性蛋白经常阻断试剂与待测物(wù)的结合。内源基质干扰物(wù)可(kě)以特异性地或非特异性地结合捕获/检测试剂或待测物(wù),使得测试信号增加或减少。
最常见的非特异性干扰是由于溶血(hemolysis)、脂肪血症(lipemia)、抗凝剂(anticoagulant)或其他(tā)小(xiǎo)分(fēn)子有(yǒu)机或无机物(wù)质的相互作用(yòng)造成的。对PK定量方法的特异性干扰可(kě)能(néng)来自蛋白药物(wù)的降解产物(wù)、内源蛋白的类似物(wù)、异质性抗體(tǐ)(heterophilic antibody)、人抗动物(wù)抗體(tǐ)、风湿因子、可(kě)溶性配體(tǐ)、抗药物(wù)抗體(tǐ)、高剂量钩效应(high-dose hook effect)等。对于潜在的干扰物(wù)通常没有(yǒu)表征良好的参照(比)物(wù)可(kě)用(yòng),而且通常不知道干扰物(wù)的性质,因而无法直接测试所有(yǒu)潜在干扰物(wù)。
传统上,会在方法开发阶段通过外加待测物(wù)/回收率实验来评估基质效应。如果制备标准曲線(xiàn)的基质与样本基质相同(例如与大多(duō)数PK定量方法一致),则二者的基质效应通常相似。优化基质的选择更多(duō)是為(wèi)了提高灵敏度(sensitivity),而不是提高准确度(absolute accuracy)。对于大多(duō)数需要替代基质的测定内源性待测物(wù)的LBA方法,需要使用(yòng)替代基质所制备的标准曲線(xiàn),计算overspiked matrix control samples中待测物(wù)的浓度,以评估绝对准确度。但是,当天然的空白基质不可(kě)及时,则需特别谨慎。由于天然基质中的内源性待测物(wù)的浓度是使用(yòng)替代基质的标准曲線(xiàn)所测定的,因此无法保证此类测定结果的绝对准确度,这是由于潜在的基质效应差异和潜在的检测方法灵敏度的限制。
平行性实验有(yǒu)助于理(lǐ)解该分(fēn)析方法的相对准确性(relative accuracy)。这是通过绘制测试信号与稀释倍数或浓度的关系图,以评估相对精度,从而评估该方法的基质效应。处理(lǐ)原始数据有(yǒu)几种不同的方法。这些方法将在"平行性、稀释線(xiàn)性和选择性"的章节中讨论。每个样本的最终曲線(xiàn)应该反映了测试试剂对待测物(wù)和基质干扰物(wù)的综合亲和力(combined binding affinity)。因此,平行性的缺失可(kě)能(néng)是基质干扰存在的标志(zhì)。
此外,通过仔细研究平行性数据的细节和趋势,方法开发人员可(kě)以发现干扰类型的相关線(xiàn)索,随后缩小(xiǎo)可(kě)能(néng)引起基质效应的物(wù)质范围。例如,如果可(kě)以通过增加稀释倍数来缓解非平行性,则可(kě)能(néng)发生了非特异性结合,可(kě)以增加该方法的MRD或优化样品稀释剂(例如添加阻滞剂、洗涤剂、盐等)来消除干扰。相反,如果不能(néng)稀释掉基质效应,则有(yǒu)可(kě)能(néng)是特异性的干扰。抑制或增强结合反应(binding reaction)可(kě)以进一步指示可(kě)能(néng)引起干扰的结合位点。可(kě)以探索识别不同表位的新(xīn)的关键试剂,增加样本预处理(lǐ)步骤(例如,碱性处理(lǐ)、酸处理(lǐ)或萃取),或添加强力洗涤剂(strong detergent),以消除此类干扰。
选择性是分(fēn)析方法的一种能(néng)力,即在样本中存在其他(tā)成分(fēn)情况下,定量地测定待测物(wù)的能(néng)力。设计良好的LBA应该能(néng)够准确地测定大多(duō)数受试者样本中的待测物(wù),而不会受到独特的个體(tǐ)基质的干扰。根据FDA指南草(cǎo)案、EMA指南和白皮书,应该外加相当于LLOQ或附近浓度的待测物(wù)到至少10个批次的人體(tǐ)个體(tǐ)基质(动物(wù)6个)中,用(yòng)以评估选择性。目前,此方法适用(yòng)于PK样本分(fēn)析方法的监管验证。
在理(lǐ)想情况下,应该使用(yòng)正常和/或患病的单个人源样本进行平行性实验。其中从可(kě)靠的来源(例如从医院获得的样本)获得的并带有(yǒu)完整的患者医疗记录和完整的样品储存记录的新(xīn)鲜样本应当得到优先考虑,或者也可(kě)以使用(yòng)从商(shāng)业来源購(gòu)买的样本,但是应考虑到所購(gòu)基质的稳定性和可(kě)靠性。
通常,稀释样本的回计算浓度可(kě)用(yòng)于评估定量分(fēn)析方法的平行性。Stevenson和Purushothama进一步建议,分(fēn)析人员可(kě)绘制稀释后调整的浓度(dilution-adjusted concentration)与多(duō)个个人样本稀释的示意图,这可(kě)以帮助设定方法的MRD并评估方法的选择性。
MRD设置应当使得大多(duō)数样品在分(fēn)析方法的定量范围内,并且保证MRD以外的多(duō)次稀释仍然能(néng)得到准确的结果。欧洲生物(wù)分(fēn)析论坛专题小(xiǎo)组总结了处理(lǐ)并行数据的替代方法。这些方法包括绘制log[微孔内测定浓度]与稀释倍数的关系图、稀释后调整的浓度与稀释倍数的关系图、稀释调整的相对误差(dilution-adjusted relative error)与稀释倍数的关系图以及log[微孔内浓度]与log[1/稀释倍数] 的关系图。这些基于回算浓度的方法允许方法开发人员根据预先设定的接受标准来评估该方法的平行性。
对相关数据进行恰当地解释数据有(yǒu)助于获取有(yǒu)利信息。例如平行性缺失可(kě)以从两个方面来解释:(1)样本之间缺乏平行性代表基质效应和试剂的选择性(选择性)问题;(2)校准品和样本之间缺乏平行性,即如果所有(yǒu)样本彼此平行但不平行于校准曲線(xiàn),则代表基质效应,或参照(比)物(wù)的特异性(specificity)问题,或两者情况都有(yǒu)。参照(比)物(wù)的特异性(specificity)问题则说明参照(比)物(wù)的质量(quality)存在问题。
图2显示了根据Stevenson和Purushothama的建议,对可(kě)溶性BCMA(sBCMA)方法案例研究数据的处理(lǐ)。在本案例研究中,连续稀释10个正常人类血清样本,使用(yòng)1%牛血清白蛋白(BSA)磷酸缓冲盐水(PBS)缓冲液,或1%BSA PBS加0.50%(v/v)Triton X-100缓冲液。校准曲線(xiàn)分(fēn)别在1%BSA PBS缓冲液以及1%BSA PBS加0.50%(v/v)Triton X-100缓冲液中制备。然后绘制稀释后调整的浓度与[1/稀释倍数]的关系图,以评估方法的平行性。
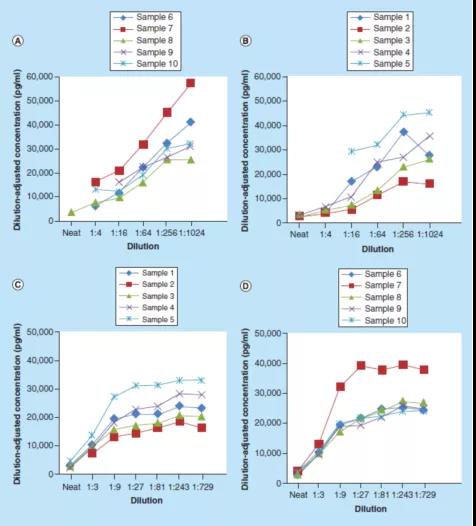
这些数据表明,使用(yòng)1% BSA PBS缓冲液作為(wèi)替代基质时,大多(duō)数受试者的样本没有(yǒu)达到平行性(图2A-B)。然而,使用(yòng)1% BSA PBS加0.50%(v/v)Triton X-100缓冲液在1:27或更大稀释倍数时,则显示了平行性(图2C-D)。
当校准曲線(xiàn)不存在或与样品不平行时,完整和稀释后样本的回算浓度要么不存在,要么不准确。所以,使用(yòng)依赖于回算浓度以评估样本之间平行性的方法可(kě)能(néng)会得出有(yǒu)误导性的结论,建议直接从分(fēn)析仪器获取原始测试信号以评估样本的平行性。
对于LBA定量方法,测试信号与浓度/稀释倍数之间的相关性通常不是線(xiàn)性的。因此,線(xiàn)性关系图不能(néng)准确反映样本之间的平行性。建议使用(yòng)加权或无加权的4或5参数logistic回归(4PL或5PL)模型,以实现最小(xiǎo)方差的最佳曲線(xiàn)拟合。从图中直接可(kě)以看到每个样本稀释曲線(xiàn)的平行关系,并可(kě)以评价样本之间的平行性。一个局限是不清楚如何為(wèi)"原始测试信号"方法设置固定的接受标准。每个样本的"B"参数("B"表示4PL和5PL的希尔曲線(xiàn)斜率)可(kě)用(yòng)于评估方法平行性。也可(kě)以进一步评估并应用(yòng)更多(duō)的统计方法,以便更好地解释数据。
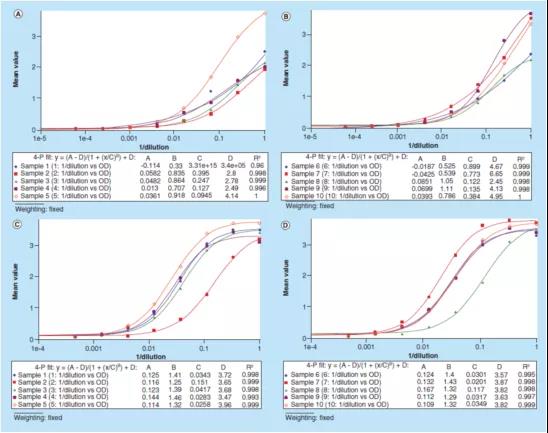
图3演示了如何使用(yòng)“原始测试信号”进行数据处理(lǐ),本图使用(yòng)了同一个sBCMA案例研究中的数据,绘制了10个样本的光密度与[1/稀释倍数]的关系图。校准曲線(xiàn)的回归模型是4PL,由测试仪器软件SoftMax® Pro完成回归分(fēn)析。实验使用(yòng)1% BSA PBS缓冲液作為(wèi)替代基质(图3A-B)时,这10个样本的B参数的范围是从0.33到1.11不等,而使用(yòng)1%BSA PBS加上0.50%(v/v)Triton X-100 缓冲液(图3C-D)作為(wèi)替代基质时,B参数的范围為(wèi)1.25到1.46。这些数据表明,在样本稀释剂(sample diluent)中加入0.50%(v/v)Triton X-100,减轻了样本的基质效应并改善了方法的平行性。
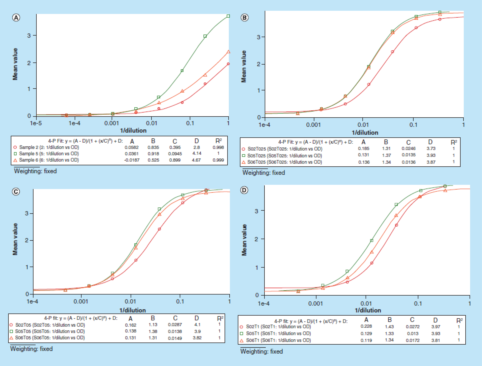
"原始信号"方法的进一步应用(yòng)是可(kě)以很(hěn)轻松地优化缓冲液,而不需要在每个不同的缓冲液中制备一条校准曲線(xiàn)。图4演示了使用(yòng)"原始测试信号"方法為(wèi)替代基质的优化,进而处理(lǐ)数据。使用(yòng)1%BSA PBS缓冲液连续稀释3个正常人血清样本,稀释剂中不含Triton X-100,然后分(fēn)别加入0.25、0.50和1.00%(v/v)的Triton X-100。所有(yǒu)稀释的样品都在同一微孔板上分(fēn)析,无需添加校准曲線(xiàn)。再利用(yòng)光學(xué)密度与[1/dilution]曲線(xiàn)关系图评价该方法的平行性。数据表明,添加了Triton X-100的3组样本其平行性都得到了提高。最终,1%BSA PBS加上0.50%(v/v)Triton X-100缓冲液被选為(wèi)替代基质,以更好地评估方法的稳健性。
由于為(wèi)"原始信号"设置固定的接受标准的方法尚不确定,因此在解决非平行性问题后,应使用(yòng)"稀释调整浓度dilution-adjusted concentration’approach "方法来确认该方法的平行性:即使用(yòng)与样本稀释缓冲液相同的缓冲液来制备校准曲線(xiàn)。
尽管其优点显而易见,但使用(yòng)平行性实验来评估方法的选择性显然也存在着挑战:并非总能(néng)获得足够多(duō)且含有(yǒu)高浓度内源性待测物(wù)的样本。另一种方法是:对多(duō)个加入标准待测物(wù)的单个样本进行稀释線(xiàn)性度实验,以评估方法的选择性。加标样品的正常浓度可(kě)设置為(wèi)内源性水平加上外加待测物(wù)的浓度。或者加标浓度可(kě)以直接用(yòng)作标称浓度,在减去内源性浓度(偏置计算bias calculation)后,可(kě)以获得正确的测定浓度。这个概念和数据评估策略应该与平行实验中所使用(yòng)的策略相同。
其他(tā)平行实验的挑战可(kě)能(néng)包括:当需要特殊基质类型(如脑脊液、组织匀浆)或基质物(wù)种(如小(xiǎo)鼠)时,有(yǒu)限的可(kě)用(yòng)小(xiǎo)样本體(tǐ)积可(kě)能(néng)不足以允许进行多(duō)次稀释后进行生物(wù)分(fēn)析。此外,没有(yǒu)统一的方法為(wèi)每个样本设置标称浓度,并评估方法的平行性。标称浓度可(kě)以设置為(wèi)来自最大稀释倍数,且其浓度高于LLOQ的MRD样本,或者是显示出平行性的所有(yǒu)稀释后浓度的平均值。也可(kě)以通过计算每个稀释后浓度与标称浓度的百分(fēn)比偏差,或者所有(yǒu)稀释后浓度的百分(fēn)比CV(%CV)评估平行性。
特异性是测试用(yòng)关键试剂(例如抗體(tǐ))區(qū)分(fēn)待测物(wù)和其他(tā)成分(fēn)的能(néng)力。LBA测量的是结合性反应活性,而不是直接测定质量。任何能(néng)够与关键试剂结合的物(wù)质都可(kě)以生成测试信号,不论其特异性和亲和力如何。这是所有(yǒu)LBA都会存在的特异性的固有(yǒu)问题。大多(duō)数LBA方法特异性测试的目标并非為(wèi)了检测绝对的特异性,而是為(wèi)达到预期的应用(yòng)目的,提供有(yǒu)关被测物(wù)的信息。
LBA方法的特异性取决于关键试剂的特异性。特异性试剂需要在其他(tā)技术(如western blot、surface plasmon resonance、HPLC、LC-MS/MS等)的协助下,进行设计、确认和选择。当前的特异性测试方案,采用(yòng)了學(xué)习-确认的方法。分(fēn)析方法的特异性是通过计算使用(yòng)外加了各种形式的潜在交叉反应物(wù)的样本的准确性来评估的。但是这是假设在干扰物(wù)质是已知的前提下,以基质或纯化物(wù)的形式而提供的。
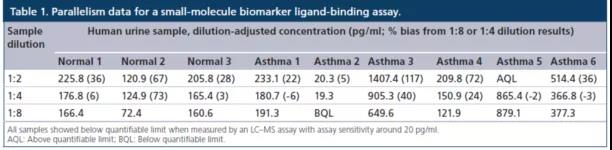
平行性实验可(kě)以间接地评估分(fēn)析方法的特异性。在方法优化后,如果在样本内以及样本和校准品曲線(xiàn)之间不能(néng)取得平行性,则关键试剂的特异性应视為(wèi)可(kě)疑。换句话说,在排除了非特异性相关的基质效应和参照(比)物(wù)的特异性问题之后,关键试剂的特异性可(kě)能(néng)是导致非平行性的主要原因。但是,与校准品曲線(xiàn)平行的样本稀释也并不能(néng)保证分(fēn)析方法的特异性。有(yǒu)这样一个例子,在一个方法比较实验中,使用(yòng)LC-MS/MS方法(灵敏度约為(wèi)20 pg/ml)来测试所有(yǒu)样品中的一个相关小(xiǎo)分(fēn)子生物(wù)标志(zhì)物(wù),检测结果是在所有(yǒu)样本均未检测到。但是,当使用(yòng)商(shāng)业ELISA试剂盒检测时,所有(yǒu)样本却均显示出可(kě)测量到的浓度,并且9个样本中有(yǒu)6个样本取得了平行性(表1)。
有(yǒu)两个与灵敏度相关的术语:LOD(limit of detection)和 LLOQ(lower limit of quantitation)。LOD是指产生与背景明显不同信号的浓度(例如背景平均值±2或3标准方差),它通常作為(wèi)灵敏度指标,供生物(wù)标志(zhì)物(wù)研究使用(yòng) (research use only,RUO)或供诊断试剂盒使用(yòng)。LLOQ是指已被证明具有(yǒu)可(kě)以接受的准确度、精密度和总误差水平的待测物(wù)的最小(xiǎo)浓度。LLOQ通常大于LOD。
传统上,生物(wù)分(fēn)析LBA方法的灵敏度(LLOQ)是在执行准确度和精密度运行时中确定的,样本是将参照(比)标准物(wù)加入到空白基质而制备的。Stevenson and和Purushothama建议分(fēn)析来自同一平行性实验的数据,但使用(yòng)校准曲線(xiàn)上的浓度(即稀释后,调整浓度之前)来确定检测内源性待测物(wù)的灵敏度。他(tā)们提出了两种不同的方法来确定LLOQ。
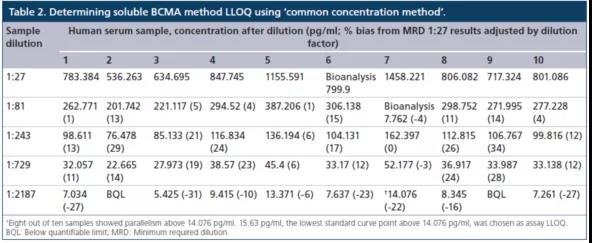
常用(yòng)稀释方法(common dilution method)是将在最大稀释倍数时给出平行响应的所有(yǒu)单个样本中观察到的最高浓度定為(wèi)LLOQ。但使用(yòng)该种办法时存在一个不足,就是当至少一个样本的内源性浓度明显高于其他(tā)样品时,确定的LLOQ将高于真正的LLOQ。而另一种方法,共同浓度方法(common concentration method)是将所有(yǒu)样品都表现出平行性的最高浓度确定為(wèi)LLOQ。换言之,任何高于确定LLOQ浓度的样本,都通过平行实验表现出相对的准确度。对sBCMA案例研究的数据,使用(yòng)了"共同浓度方法"(表2)。
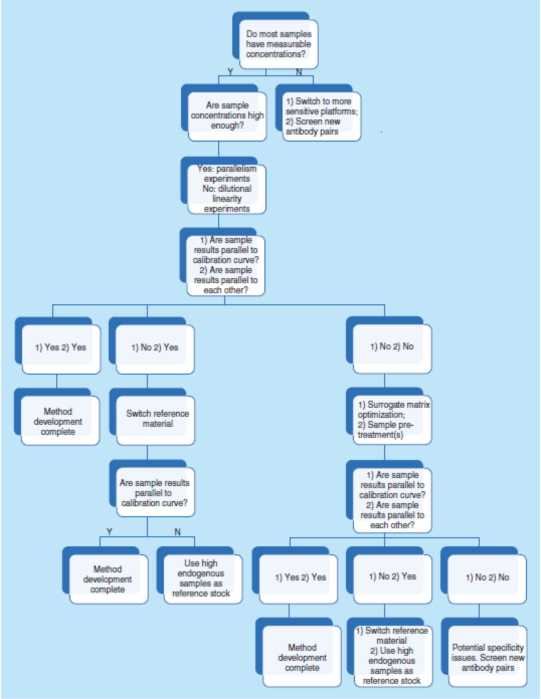
总之,平行实验是指导早期方法开发和优化的有(yǒu)力工具。它直接评估待测物(wù)的内源性浓度、测定基质效应、选择性和灵敏度,它也是方法特异性的一个间接指标。图5显示了一个使用(yòng)平行数据进行系统性方法开发和优化的策略决策树。
可(kě)按质量级别(quality level)或预期用(yòng)途对商(shāng)用(yòng)试剂盒进行分(fēn)类。无论是用(yòng)于化验室测试的商(shāng)业试剂盒(以支持临床诊断或预后),还是用(yòng)于支持药物(wù)开发的生物(wù)分(fēn)析测试,方法开发的目标都是一样的,即建立一个有(yǒu)足够准确度和灵敏度的、符合预期用(yòng)途的、可(kě)靠的定量分(fēn)析方法。
生物(wù)分(fēn)析业界讨论了使用(yòng)商(shāng)业试剂盒开发和验证定量分(fēn)析方法的策略,FDA指南草(cǎo)案和EMA指南指出:需要重新(xīn)验证用(yòng)于药物(wù)开发目的的商(shāng)业试剂盒,以确保其可(kě)靠性。业界中不同的组织在相关综述或研究论文(wén)中总结了当前商(shāng)业试剂盒所面临的挑战以及关于如何采纳和调整商(shāng)业试剂盒的建议。也有(yǒu)相关白皮书讨论了使用(yòng)singleplex and multiplex试剂盒的方法的验证建议。试剂盒的最终用(yòng)户还发表了一些案例研究,展示了他(tā)们采用(yòng)和认证研究级别试剂盒的策略,这些试剂盒来自不同的供应商(shāng),并且用(yòng)于不同的目的。详见文(wén)末的参考文(wén)献。
简而言之,需要确认参考照(比)物(wù)料和关键试剂的质量和批次间的变异性,也需要确认或重新(xīn)建立测试方法的LLOQ和MRD,也可(kě)能(néng)需要优化缓冲液和稀释剂,并且在有(yǒu)必要时优化检测步骤。而评估上述所有(yǒu)内容最有(yǒu)效的方法则是通过平行性实验。因此,强烈建议在方法开发的早期阶段进行平行性实验。如果在研究期间观察到非平行性,则建议使用(yòng)相同的故障排除路線(xiàn)(见图5)。
与单通路LBAs相比,且考虑到节省样本體(tǐ)积和分(fēn)析时间,多(duō)通路复用(yòng)的LBAs似乎更具吸引力。但是,由于检测环境极其复杂,大多(duō)数多(duō)通路复用(yòng)测试方法暂无法达到与单通路测试方法相同的质量水平。
关于符合其用(yòng)途(fit-for-purpose)的多(duō)通路复用(yòng)LBA方法的验证,Jani等人指出,除了面临与单通路LBA方法相同的挑战外,对于多(duō)通路复用(yòng)LBA方法的开发还需要考虑其它独特的挑战: 包括所有(yǒu)待测物(wù)的定量范围和MRD的设置,不同试剂抗體(tǐ)对和待测物(wù)之间的交叉反应(cross-reactivity)以及串扰(crosstalk),由于well-to-well or spot-to-spot physical‘carryover残留’所造成的串扰。
平行性研究(parallelism)或外加待测物(wù)的稀释線(xiàn)性度(spiked dilutional linearity)实验可(kě)以应对上面所列举的挑战: (1)灵敏度和MRD:可(kě)以使用(yòng)与单通路LBA相同的方法,对每个待测物(wù)设置灵敏度要求,应当选择对所有(yǒu)待测物(wù)实现平行性的最大稀释度作為(wèi)方法的MRD;(2)交叉反应:可(kě)以通过将不同浓度的捕获或检测抗體(tǐ)分(fēn)别加入到不同的样本中,然后进行平行性实验来评估;(3)串扰(Crosstalk):可(kě)以通过将每个高浓度待测物(wù)分(fēn)别外加到不同的样本中,然后进行稀释線(xiàn)性度(spiked dilutional linearity/spiked parallelism)实验来评估,或者可(kě)以选择一个样本,其某个待测物(wù)的内源性浓度显著地高于其他(tā)待测物(wù)(如果有(yǒu)的话),然后进行平行性实验。
当然还有(yǒu)其他(tā)办法可(kě)以解决交叉反应和串扰的问题。如“失踪的人(Missing man)技术”,即除一种试剂外,每次将所有(yǒu)关键试剂均添加到测试中,可(kě)用(yòng)于评估试剂的交叉反应。此外,改变一个待测物(wù)的浓度,同时保持其它待测物(wù)处于低浓度范围,是解决串扰问题的另一个方法。如果使用(yòng)商(shāng)业试剂盒,会由于重复这些实验而造成较高的复杂性和成本,如果有(yǒu)可(kě)能(néng)的话建议始终从试剂盒生产商(shāng)处获取相关原始数据的副本。
平行性实验可(kě)用(yòng)于研究LBA分(fēn)析方法的基质效应、选择性和灵敏度等问题。然而,作為(wèi)所有(yǒu)LBA方法都存在的问题,并不能(néng)直接通过平行性实验来研究方法的特异性(specificity)。但在方法优化之后,若样本分(fēn)析中出现了平行性的缺失,则很(hěn)可(kě)能(néng)是由于其特异性不佳。
LBA方法的特异性(specificity)取决于关键的测试试剂(例如,抗體(tǐ)对)。关键试剂的特异性必须由供应商(shāng)或最终用(yòng)户进行评估,以确保方法的效能(néng)。平行性数据的数學(xué)解释為(wèi)表征LBA方法的特异性提供了一些線(xiàn)索。此外,还可(kě)以评估和使用(yòng)更多(duō)的数學(xué)或统计工具,以便从平行性数据中找出更多(duō)细节(关于试剂结合待测物(wù)、干扰物(wù)的活性)。其他(tā)技术如western blot,surface plasmon resonance,HPLC和LC-MS/MS,也可(kě)助于确定非平行性的根本原因,分(fēn)析人员应根据需要适当使用(yòng)这些技术。
然而,对用(yòng)于内源性待测物(wù)的LBA方法,尚不清楚特异性表征到什么程度才是足够的。且如果要确定绝对的特异性(事实上并不总是需要),就需要投入大量的资源,而这些资源本可(kě)以在其它地方使用(yòng),因為(wèi)开发具有(yǒu)良好特征的内源性LBA的最终目标是确保它们具有(yǒu)足够的灵敏度和准确度,以定量分(fēn)析想要测量的变化和物(wù)质。
平行性研究是通过评估稀释对生物(wù)基质中待测物(wù)定量分(fēn)析的影响,来表述相对准确性的一项关键指标。可(kě)以评估的相关分(fēn)析方法的基本特征包括选择性、基质效应、所需最低稀释倍数、健康和患病人群中内源性待测物(wù)的浓度和LLOQ。
下面将简单介绍对于这类LBA方法应如何进行类似的评估。
表3显示了使用(yòng)平行性、稀释線(xiàn)性度(加标平行性)和加标/回收方法来评估内源性LBA方法关键参数的优缺点。与大多(duō)数外源性药物(wù)的定量方法不同,内源性(例如,生物(wù)标志(zhì)物(wù))LBA方法通常需要替代参照(比)标准物(wù)和不含有(yǒu)待测物(wù)的替代基质。用(yòng)于内源性LBA的照(比)标准物(wù),通常既非高度纯化,也非表征良好,且很(hěn)可(kě)能(néng)不能(néng)完全代表待测的内源性蛋白质,这些蛋白质通常是异质性(heterogeneous)的,在其天然基质中存在着多(duō)个异构體(tǐ)(isoforms)。
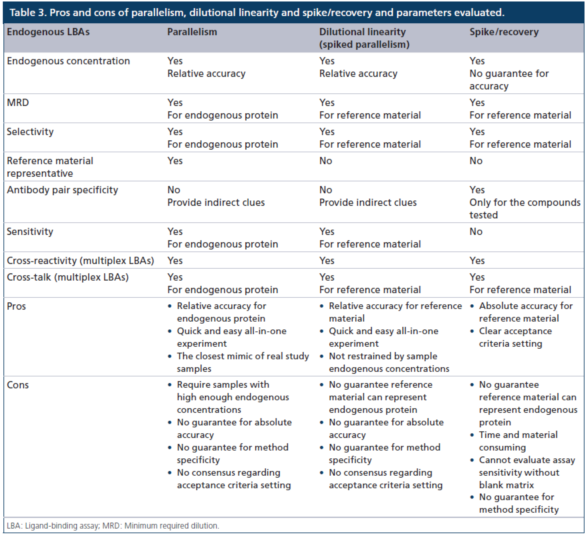
替代基质的基质效应由于基质生物(wù)學(xué)的不同,通常会不同于正常人或目标疾病人群的基质。用(yòng)于内源性待测物(wù)的LBA方法的所有(yǒu)独特特性使得这样的分(fēn)析方法往往难于使用(yòng)与传统的完全定量的PK方法相同的方式去进行开发和评估。开发具有(yǒu)良好特征的、相对定量的LBA方法的首要目标是區(qū)分(fēn)疾病和正常人群(即以诊断/预后為(wèi)目的),或區(qū)分(fēn)来自经药物(wù)治疗/未经治疗的个别样本(即以药物(wù)开发為(wèi)目的),而不是测定每个样本的实际真实浓度。
加标/回收实验被证明是用(yòng)于完全定量PK的LBA方法的开发和验证最有(yǒu)效的和应用(yòng)广泛的方法,但相对精度的测试方法并不总是需要做这些实验。当样本没有(yǒu)足够高的内源性待测物(wù)浓度以支持平行性实验时,加标稀释線(xiàn)性度的实验可(kě)用(yòng)于证明分(fēn)析方法的相对准确度。需要仔细测试外加的参照(比)标准物(wù)和内源性天然蛋白之间的差别。
尽管生物(wù)分(fēn)析业界就如何进行平行性实验以及如何评估平行性数据尚未达成完全的共识,但这不影响将平行性数据用(yòng)于方法开发和优化。平行性实验是一个强大的工具,可(kě)以解决用(yòng)于内源性待测物(wù)定量的LBA方法的基本问题(方法的特异性除外)。平行性实验也提供了对最接近实际研究样本的模仿,并且為(wèi)相对定量的和适合其用(yòng)途(fit-for-purpose)的分(fēn)析方法的设计提供了充分(fēn)的信息。
本文(wén)如有(yǒu)疏漏和误读相关指南、数据的地方,请读者评论和指正。所有(yǒu)引用(yòng)的原始信息和资料均来自已经发表學(xué)术期刊、官方网络报道等公开渠道, 不涉及任何保密信息。参考文(wén)献的选择考虑到多(duō)样化但也不可(kě)能(néng)完备。欢迎读者提供有(yǒu)价值的文(wén)献及其评估。

1. Plikaytis BD, et al. Determination of parallelism and nonparallelism in bioassay dilution curves. J. Clin. Microbiol. 32, 2441–2447 (1994).
2. Draft Guidance for Industry, Bioanalytical Method Validation. US Department of Health and Human Services, US FDA, Center for Drug Evaluation and Research, Center for Veterinary Medicine, MD, USA 2013).www.fda.gov/downloads/drugs/guidance compliance
3. Tu, J. and P. Bennett, Parallelism experiments to evaluate matrix effects, selectivity and sensitivity in ligand-binding assay method development: pros and cons. Bioanalysis, 2017. 9(14): p. 1107-1122.
4. Guideline on Bioanalytical Method Validation. European Medicines Agency, London, UK. (2011).www.ema.europa.eu/docs/en_GB/document library/
5. DeSilva B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm. Res. 20(11), 1885–1900 (2003).
6. Stevenson LF, Purushothama S. Parallelism: considerations for the development, validation and implementation of PK and biomarker ligand-binding assays. Bioanalysis 6(2), 185–198 (2014).
7. Lee JW, et al. Bioanalytical approaches to quantify ‘total’ and ‘free’ therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J. 13(1), 99–110 (2013).
8. DeSilva B, et al. 2012 White paper on recent issues in bioanalysis and alignment of multiple guidelines. Bioanalysis 4(18) 2213–2226 (2012).
9. Stevenson L, et al. Large molecule specific assay operation: recommendations for the best practices and harmonization from the global bioanalysis consortium harmonization team. AAPS J. 16(1), 83–88 (2013).
10. Booth B, et al. Workshop report: Crystal City V-quantitative bioanalytical method validation and implementation: the 2013 revised FDA guidance. AAPS J. 17(2), 277–288 (2015).
11. Tate J, Ward G. Interferences in immunoassay. Clin. Biochem. Rev. 25(2), 105–120 (2004).
12. Kricka LJ. Human anti-animal antibody interferences in immunological assays. Clin. Chem. 45(7), 942–956 (1999).
13. Lee J, Ma H. Specificity and selectivity evaluation of ligand binding assay of protein therapeutics against concomitant drugs and related endogenous proteins. AAPS J. 9(2) E164–E170 (2007).
14. Gorovits B, et al. Protein-based matrix interferences in ligand-binding assays. Bioanalysis 6(8),1131–1140 (2014).
15. Hennig C, et al. The influence of naturally occurring heterophilic anti-immunoglobulin antibodies on direct measurement of serum protein using sandwich ELISAs. J. Immunol. Methods 235(1–2), 71–80 (2000).
16. Schwichart M, et al. Interference in immunoassays to support therapeutic antibody development in preclinical and clinical studies. Bioanalysis 6(14),1939–1951 (2014).
17. Salimi-Moosavi H, et al. Novel approaches using alkaline or acid/guanidine treatment to eliminate therapeutic antibody interference in the measurement of total target ligand. J. Pharm. Biomed. Anal. 51(5), 1128–1133 (2010).
18. Bastarache JA, et al. Accuracy and reproducibility of a multiplex immunoassay platform: a validation study. J. Immunol. Methods 367(1–2), 33–39 (2011).
19. Stevenson L, et al. Large molecule specific assay operations: recommendations for best practices and harmonization from the Global Bioanalysis Consortium Harmonization Team. AAPS J. 16(1), 83–88 (2014).
20. Jani D, et al. Recommendations for use and fit-for-purpose validation of biomarker multiplex ligand binding assays in drug development. AAPS J. 18(1), 1–14 (2016).
21. Bowsher RR, Sailstad JM. Insights in the application of research-grade diagnostic kits for biomarker assessments in support of clinical drug development: bioanalysis of circulating concentrations of soluble receptor activator of nuclear factor κβ ligand. J. Pharm. Biomed. Anal. 48(5), 1282–1289 (2008).
22. Nowatzke W, et al. Systematic analytical validation of commercial kits for the determination of novel biomarkers for clinical drug development. Bioanalysis 2(2), 237–247 (2010).
23. Ray CA, et al. Development, validation, and implementation of a multiplex immunoassay for the simultaneous determination of five cytokines in human serum. J. Pharm. Biomed. Anal. 36(5), 1037–1044 (2005).
24. Aldo P, et al. Simple Plex™: a novel multi-analyte, automated microfluidic immunoassay platform for the detection of human and mouse cytokines and chemokines. Am. J. Reprod. Immunol. 75(6), 678–693 (2016).












