









日前,博济医药董事長(cháng)王廷春作客“博济研语”直播间,以《上市许可(kě)持有(yǒu)人的前世今生》為(wèi)题,向广大网友全面介绍了药品上市许可(kě)持有(yǒu)人制度的来龙去脉。
鉴于广大网友对直播报道中的快问快答(dá)环节颇為(wèi)喜欢,此次直播报道将全程采用(yòng)快问快答(dá),给广大网友在阅读上以全新(xīn)體(tǐ)验。
MAH与生产企业、经营企业、CRO、原辅料供应商(shāng)等是合同关系;
MAH是全生命周期的责任主體(tǐ),依据产品质量法和侵权法等承担产品责任;
MAH必须通过合同约定、保险和基金分(fēn)担风险;
MAH可(kě)以在任意环节任意选择合作伙伴,但是不管选择什么,主要责任集中到MAH。
“要想弄清楚MAH制度的活力优势,首先要弄清楚在没有(yǒu)MAH制度前,整个行业运行过程中的遇到了哪些问题。”在解读MAH制度意义之前,王廷春首先分(fēn)析了没有(yǒu)实施MAH制度前,医药产业创新(xīn)发展的瓶颈所在。

“通过MAH制度可(kě)以鼓励药物(wù)研发创新(xīn),加快创新(xīn)药企研发成果转换,打破了“卖青苗”窘境,药品研发机构、科(kē)研人员以及药品生产企业的研发热情被进一步调动。”王廷春说,MAH制度的实施还有(yǒu)效遏制低水平重复建设、品质落后、品种重复,产能(néng)浪费,同时节约了资源,进一步在法律层面明确了药品核心权益归属权,有(yǒu)利于整个生产行业水平提高,改变生物(wù)医药行业研发、生产、销售等各领域企业关系与市场生态结构。
不具备药品生产资格的研发企业、以研发能(néng)力為(wèi)核心的创新(xīn)药海归药企、需要战略转型的大中型医药企业、具有(yǒu)创新(xīn)能(néng)力和创新(xīn)产品的CRO等第三方研发外包企业(包括:CRO、CMO、CDMO等)
作為(wèi)深耕CRO行业多(duō)年的专家,王廷春给出了自己的答(dá)案。


王廷春说,按照规定申请MAH药品不限种类,國(guó)内外的化药、中药、生物(wù)制品均可(kě)进行申报,但需要说明的是,毒麻精放类和疫苗类药物(wù)申请人不得委托生产。
在王廷春看来,成為(wèi)MAH需要在内外部两方面具备一定的条件。
从内部上看,必须具备质量管理(lǐ)能(néng)力、风险防控能(néng)力、责任赔偿能(néng)力。王廷春进一步解释道:“首先,持有(yǒu)人必须建立一套药品全生命周期全过程的质量管理(lǐ)體(tǐ)系,并且要有(yǒu)相应技能(néng)的管理(lǐ)人才,确保质量管理(lǐ)體(tǐ)系的正常运行。同时,持有(yǒu)人具备风险识别、风险预警、风险消除、偏差处理(lǐ)的能(néng)力,做好全生命周期的药物(wù)警戒管理(lǐ)。此外,持有(yǒu)人还要具备责任赔偿能(néng)力。”
从外部条件上看,拥有(yǒu)自主知识产权、能(néng)够独立承担责任、申请地具有(yǒu)中國(guó)國(guó)籍的科(kē)研人员(法人是中國(guó)公民(mín))的药品研发机构或企业(注册地不受限制,境外企业需在境内注册企业履行相关义務(wù))是成為(wèi)持有(yǒu)人的关键要素。
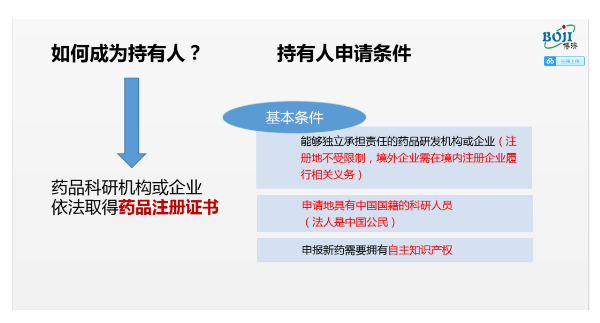

“根据新(xīn)版《药品管理(lǐ)法》规定,药品上市许可(kě)持有(yǒu)人是指取得药品注册证书的企业或者药品研制机构等。”
在王廷春看来,一个等字用(yòng)意颇深。“在2015年有(yǒu)关部门首次提出MAH制度时,并未就个人成為(wèi)持有(yǒu)人予以否定,但在新(xīn)版《药品管理(lǐ)法》中个人成為(wèi)持有(yǒu)人的内容却未被提及。”王廷春建议,个人可(kě)通过成立研发机构或公司的方式成為(wèi)持有(yǒu)人。

“针对于新(xīn)注册药品、已批准上市药品、此外在变更申请、其它要求也都有(yǒu)着明确要求。”王廷春说。
如果境外上市许可(kě)持有(yǒu)人生产在境外,销售在境内,由其指定的在中國(guó)境内的企业法人履行药品上市许可(kě)持有(yǒu)人义務(wù),与药品上市许可(kě)持有(yǒu)人承担连带责任,可(kě)以依法取得我國(guó)药品经营许可(kě)证。
國(guó)外的药品研发机构申请我國(guó)的药品上市许可(kě)持有(yǒu)人,法律没有(yǒu)明确规定。从一般法理(lǐ)来看,应当依法成為(wèi)我國(guó)的药品上市许可(kě)持有(yǒu)人,但其不需要取得药品生产许可(kě)证。
“新(xīn)版《药品管理(lǐ)法》没有(yǒu)禁止境外的药品上市许可(kě)持有(yǒu)人委托境内受托生产企业进行生产,境内的药品上市许可(kě)持有(yǒu)人委托境外受托生产企业应当是符合条件的药品生产企业,但是并没有(yǒu)要求受托生产企业必须取得《药品生产企业许可(kě)证》。”王廷春说,如果允许跨境委托生产,将对现行的药品注册和管理(lǐ)體(tǐ)系带来巨大的挑战,从而可(kě)能(néng)推动进一步的制度改革。

“办理(lǐ)药品生产许可(kě)证大致分(fēn)為(wèi)申请类型、取得条件、取得方法、办理(lǐ)程序时限、现场核查要求等步骤。”在王廷春看来,现场核查是尤為(wèi)重要的一步,持有(yǒu)人在责任赔偿方面的抗压能(néng)力或将成為(wèi)核查的关键内容。
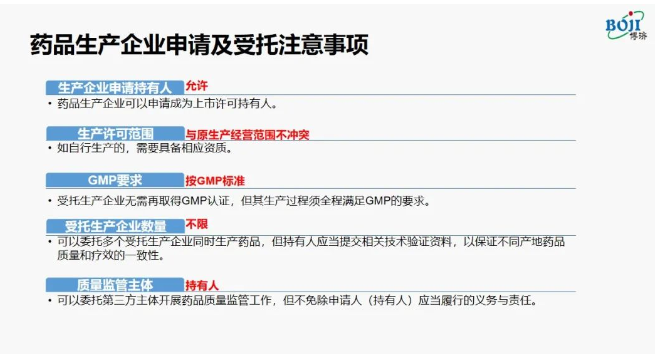
受托生产企业必须是中國(guó)境内所在行政區(qū)域内依法设立、持有(yǒu)相应药品生产范围的《药品生产许可(kě)证》、符合GMP生产质量管理(lǐ)规范。
临床试验用(yòng)药物(wù):生产单位可(kě)以是尚未取得相应范围《药品生产许可(kě)证》或者《药品生产质量管理(lǐ)规范》认证证书的单位,但制备过程应当严格执行GMP的有(yǒu)关要求。
此外受托企业要明确自身的义務(wù)和责任,要严格履行《药品管理(lǐ)法》以及其他(tā)法规规定的有(yǒu)关药品生产企业在药品生产方面的义務(wù),并且承担相应的法律责任。同时要履行与持有(yǒu)人依法约定的相关义務(wù),并且承担相应的法律责任。



在2019年12月1日前,药品上市许可(kě)持有(yǒu)人与受托药品生产企业已签订的委托销售合同,在合同期间内受托药品生产企业可(kě)继续销售药品,合同到期后不得继续委托药品生产企业销售药品(原则上,药品上市许可(kě)持有(yǒu)人委托药品生产企业销售药品不得超过2022年12月31日)。
2019年12月1日后,药品上市许可(kě)持有(yǒu)人不得与受托药品生产企业签订委托销售合同,签订合同销售的,责令限期整改;逾期不改正的,依据《药品管理(lǐ)法》第一百一十五条处理(lǐ)。














