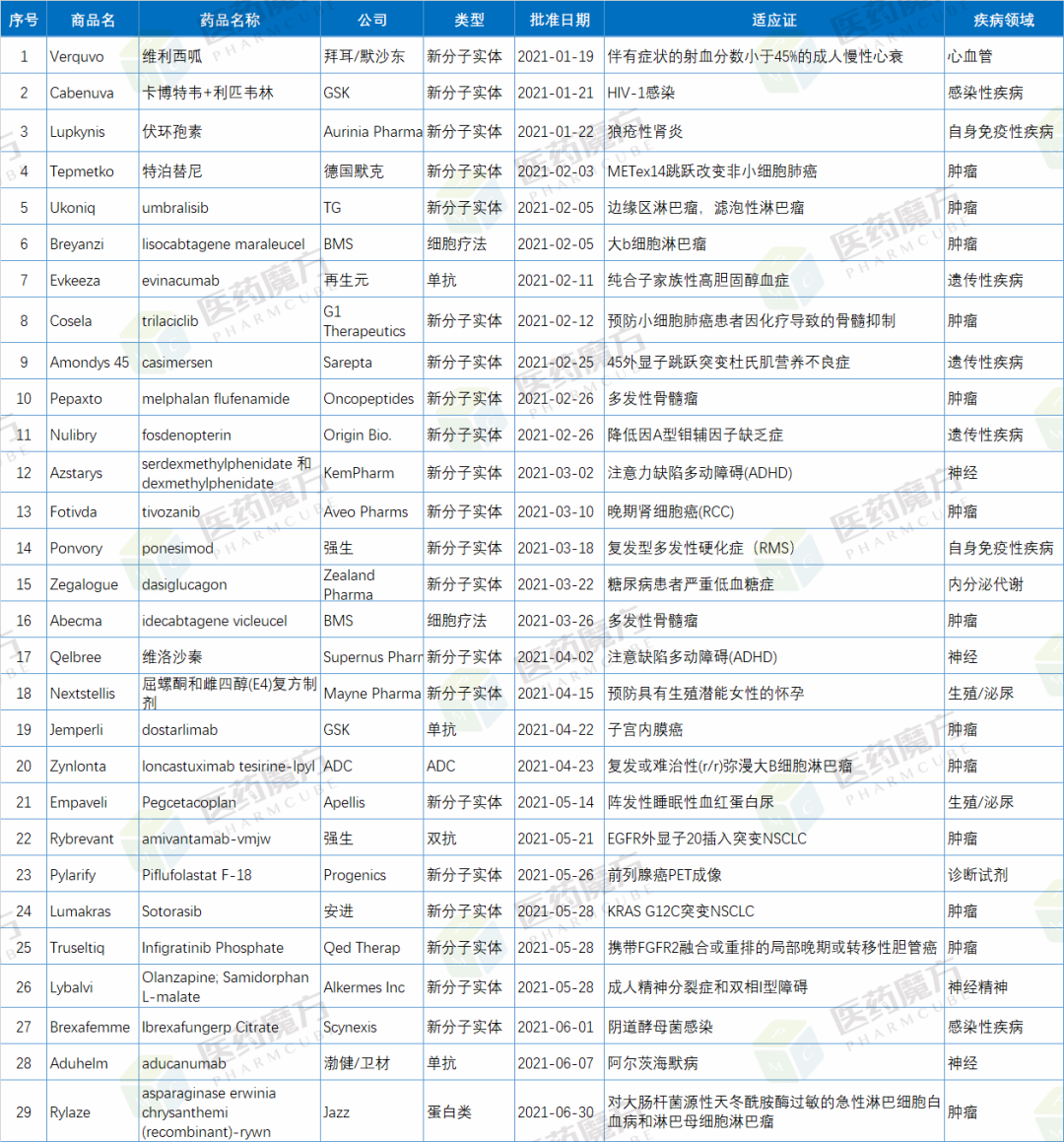
2021年上半年,FDA共批准27款新(xīn)药,其中包括21款新(xīn)分(fēn)子实體(tǐ)药物(wù),6款生物(wù)制品。另外,FDA还批准了2款细胞疗法。

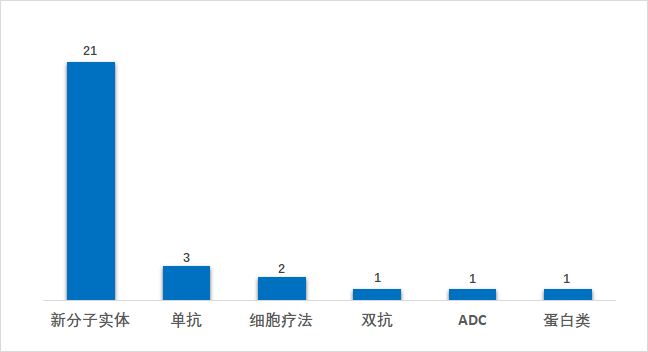
从疾病领域来看,2021年上半年FDA批准的新(xīn)药仍以肿瘤药居多(duō),占比45%(13/29),神经精神(14%,4/29)、遗传性疾病(10%,3/29)、生殖泌尿系统疾病7%(2/29)、感染性疾病7%(2/29)等领域也分(fēn)别有(yǒu)新(xīn)药获批。
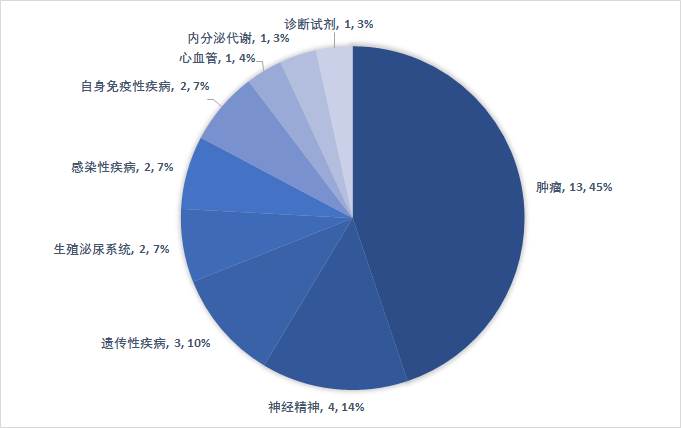
从审评方式上看,上半年共有(yǒu)20个(69%)新(xīn)药是以“优先审评”的方式获得FDA批准上市,包括14款新(xīn)分(fēn)子实體(tǐ)、和6款生物(wù)药。有(yǒu)11个品种被FDA授予“孤儿药”资格,占比38%。
上半年获批的新(xīn)药中,有(yǒu)9款属于First-in-class疗法,具體(tǐ)品种如下:
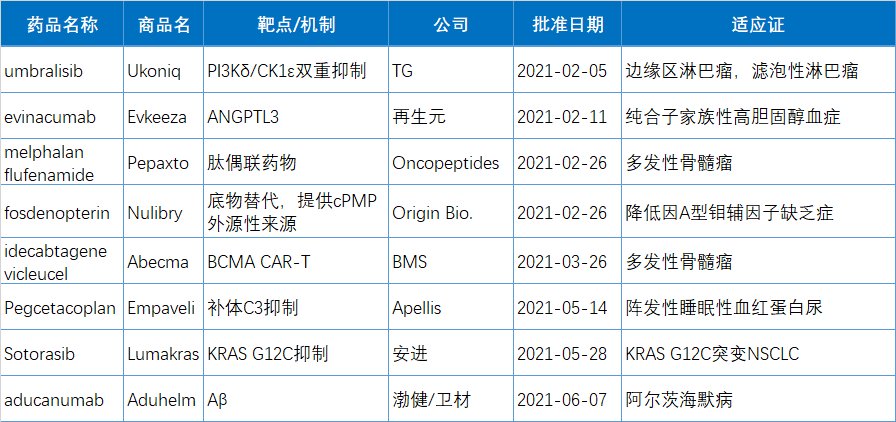
上半年有(yǒu)10款新(xīn)药(34.5%)被FDA授予突破性疗法(BTD)资格,包括5个生物(wù)制品,占上半年获批生物(wù)制品的63%。另有(yǒu)5个获得BTD资格的是小(xiǎo)分(fēn)子药物(wù),占到今年获批小(xiǎo)分(fēn)子药物(wù)的24%。
以下為(wèi)10款突破性疗法简单介绍:
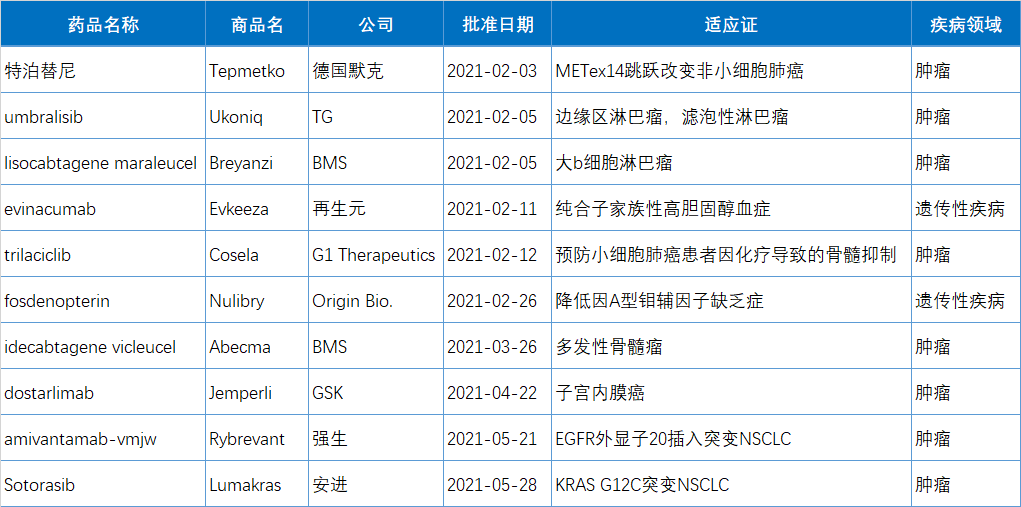
特泊替尼(tepotinib)
适应症:治疗携带MET基因第14号外显子(MET ex14)跳跃突变的转移性非小(xiǎo)细胞肺癌(NSCLC)成人患者,这类侵袭性肺癌患者通常是老年人,预后较差。目前临床迫切需要能(néng)够产生持久抗肿瘤活性的靶向疗法以改善这种挑战性疾病患者的生活。
2月3日,德國(guó)默克(Merck KGaA)旗下EMD Serono宣布,FDA已加速批准口服MET抑制剂特泊替尼(tepotinib,EMD 1214063,Tepmetko)上市,用(yòng)于治疗携带MET外显子14(METex14)跳跃变异的转移性非小(xiǎo)细胞肺癌(NSCLC)成人患者。
特泊替尼(tepotinib)是全球上市的第一款MET抑制剂(于2020年3月由日本厚生劳动省首先批准上市),FDA批准的第二款MET抑制(第一款為(wèi)卡马替尼);同时是FAD批准的第63个小(xiǎo)分(fēn)子激酶抑制剂,也是FDA在2021年批准的第1个小(xiǎo)分(fēn)子激酶抑制剂。
这一批准基于一项有(yǒu)152名携带METex14跳跃变异的晚期或转移性NSCLC患者参与VISION 2期临床试验,验结果显示:替波替尼(tepotinib)在初治和经治患者中的总缓解率均达到43%。初治和经治患者的中位缓解持续时间(DOR)分(fēn)别為(wèi)10.8个月(95% CI,6.9-NE)和11.1个月(95% CI,9.5-18.5)。67%的初治患者和75%的经治患者的缓解持续时间為(wèi)6个月以上,30%的初治患者和50%的经治患者的缓解持续时间為(wèi)9个月以上。
特泊替尼(tepotinib)是默克内部发现的一种口服MET激酶抑制剂,可(kě)强效、高度选择性抑制由MET(基因)改变——包括MET第14号外显子跳跃突变、MET扩增或MET蛋白过表达引起的致癌信号,具有(yǒu)改善携带这些特定MET改变的侵袭性肿瘤患者治疗预后的潜力。除了NSCLC之外,默克也正在积极评估特泊替尼(tepotinib)联合新(xīn)疗法治疗其他(tā)肿瘤适应症。
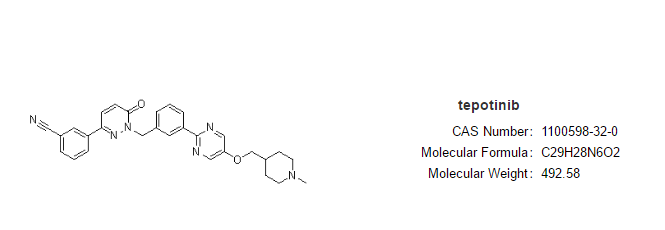
umbralisib
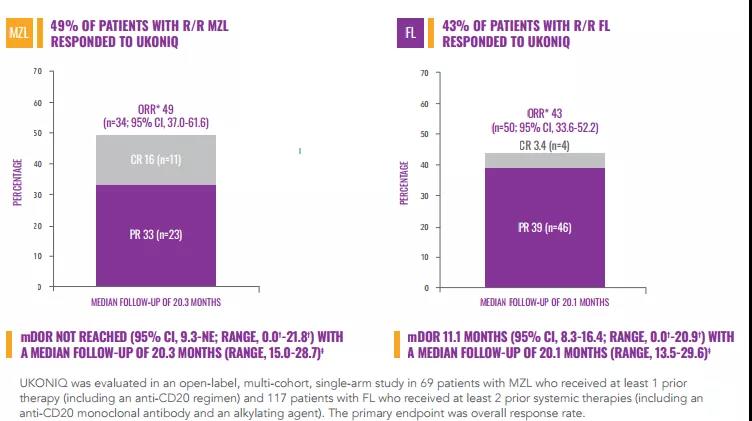
适应症:用(yòng)于治疗之前至少接受过一种基于抗CD20治疗方案的复发或难治性边缘區(qū)淋巴瘤(MZL)成人患者和之前至少接受过三線(xiàn)全身治疗的复发或难治性滤泡性淋巴瘤(FL)成人患者。
Umbralisib由TG公司开发,是首个也是唯一一个获批上市的每日口服1次的磷酸肌醇3激酶(PI3K ) δ和酪蛋白激酶1 (CK1) ε抑制剂。PI3K-δ在支持细胞增殖和生存、细胞分(fēn)化、细胞间运输和免疫方面发挥重要作用(yòng),在正常和恶性b细胞中都有(yǒu)表达。CK1-ε是一种癌蛋白翻译调节因子,与癌细胞包括淋巴恶性肿瘤发病机制有(yǒu)关。此前,umbralisib治疗MZL适应症曾获FDA突破性疗法认定,上市申请也被纳入优先审评。umbralisib也被授予治疗MZL和FL的孤儿药认定 (ODD)。
umbralisib分(fēn)子结构式
MZL和FL适应症的加速获批是基于一项II期UNITY-NHL (NCT02793583)研究的总缓解率(ORR)数据。UNITY-NHL研究是一项开放标签,多(duō)中心,双队列研究,2个队列分(fēn)别招募了69例 MZL患者和117 例FL患者,受试者接受每日口服1次umbralisib(800mg)治疗,直至疾病进展或不可(kě)耐受毒性。研究结果表明,MZL患者的总缓解率(ORR)為(wèi)49% ,完全缓解率(CR)為(wèi)16%,中位缓解持续时间(DOR)尚未达到。FL患者ORR為(wèi)43%,CR為(wèi)3.4%,DOR為(wèi)11.1个月。
lisocabtagene maraleucel
适应症:治疗成人复发或难治性大b细胞淋巴瘤。
2021年2月5日,FDA批准了lisocabtagene maraleucel用(yòng)于经≥2線(xiàn)全身治疗的复发/难治性(R/R)大B细胞淋巴瘤成人患者,包括弥漫性大B细胞淋巴瘤(DLBCL)、高级别B细胞淋巴瘤、原发性纵隔大B细胞淋巴瘤和3B级滤泡性淋巴瘤患者。
在192例可(kě)评估疗效的患者中,根据独立审查委员会(IRC)评估的总體(tǐ)缓解率(ORR)為(wèi)73%(95%CI:67-80),完全缓解(CR)率為(wèi)54%(95%CI:47-61)。至第一次缓解的中位时间為(wèi)1个月。在104例达到CR的患者中,有(yǒu)65%的患者缓解持续了至少6个月,62%的患者缓解持续了至少9个月。CR患者的估计中位缓解持续时间(DOR)未达到(95%CI:16.7-NR),部分(fēn)缓解(PR)患者的估计中位DOR為(wèi)1.4个月(95%CI:1.1-2.2)。
evinacumab
适应症:作為(wèi)其他(tā)降脂药物(wù)的辅助疗法用(yòng)于治疗成人和12岁以上儿科(kē)纯合子家族性高胆固醇血症(HoFH)。
Evinacumab是一种靶向血管生成素样蛋白3 (ANGPTL3) 的全人源IgG4κ型单抗,是一种全新(xīn)机制的降脂药,通过抑制ANGPTL3而间接加速體(tǐ)内脂肪的降解。此前已获得FDA授予的孤儿药资格,优先审评资格和突破性疗法认定,是第一个上市的ANGPTL3抑制剂。
HoFH是一种罕见的家族遗传性脂质代謝(xiè)异常疾病,病因是控制人體(tǐ)清除胆固醇的PCSK9,LDLR或APOB基因在复制时产生突变,导致胆固醇无法正常代謝(xiè)。患者通常伴随着极高的低密度脂蛋白 (LDL-C)水平,通常在青少年期就发展為(wèi)严重的心血管疾病,且治疗方案有(yǒu)限,常规的PCSK9抑制剂和其他(tā)降脂疗法(如他(tā)汀类药物(wù))对其都没有(yǒu)显著疗效,此次evinacumab的获批,可(kě)谓是HoFH患者期盼已久的福音。
trilaciclib
适应症:预防扩散期小(xiǎo)细胞肺癌成人患者因铂类╱依托泊苷方案或拓扑替康方案化學(xué)治疗导致的骨髓抑制。
G1 Therapeutics于今年2月底对外宣布,FDA已批准Cosela(trilaciclib)注射液,用(yòng)于广泛期非小(xiǎo)细胞肺癌(ES-SCLC)成人患者,在接受含铂/依托泊苷的化疗方案或含拓扑替康的化疗方案之前给药,以降低化疗诱导的骨髓抑制的发生率。Cosela通过优先审查程序获得批准,之前已被美國(guó)FDA授予突破性药物(wù)资格(BTD)。
值得一提的是,Cosela是全球第一款也是唯一一款可(kě)降低化疗诱导的骨髓抑制发生率的骨髓保护疗法,能(néng)够為(wèi)接受化疗的ES-SCLC患者骨髓起到保护作用(yòng)。Cosela的骨髓保护作用(yòng),可(kě)降低严重中性粒细胞减少症和贫血的发生率和持续时间、减少诸如生長(cháng)因子和红细胞输注等抢救性干预措施的需要。红细胞输注等抢救性干预措施的需要。
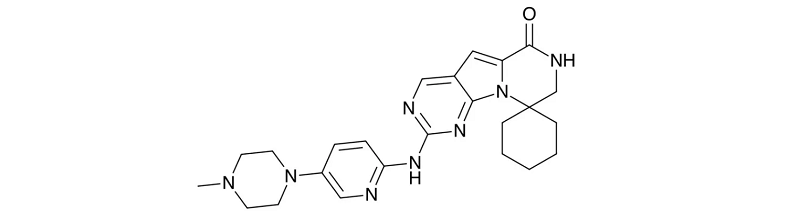
fosdenopterin
适应症:用(yòng)于降低因A型钼辅因子缺乏(MoCD)导致的死亡风险。
Nulibry(fosdenopterin)由BridgeBio子公司Origin Biosciences开发,是FDA批准的首款治疗该疾病创新(xīn)疗法。A型钼辅因子缺乏症是一种罕见的、遗传代謝(xiè)性疾病,通常发病于出生几日的婴儿,会导致顽固性癫痫发作、脑损伤和死亡。全球共不到150例患者受此疾病影响,患者中位生存时间為(wèi)4年。
Fosdenopterin是一种底物(wù)替代疗法,提供了cPMP(环吡喃单磷酸)的外源性来源,cPMP可(kě)转化為(wèi)钼嘌呤,而钼嘌呤又(yòu)可(kě)转化為(wèi)钼辅因子,以防止因钼辅因子缺乏而导致的亚硫酸盐氧化酶合成减少进而导致的毒亚硫酸盐的积累,从而缓解婴幼儿A 型MoCD 的中枢神经系统症状。Nulibry曾获得FDA授予的优先审评资格、突破性疗法认定和孤儿药资格认定。
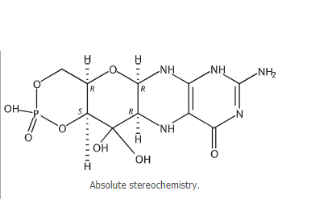
idecabtagene vicleucel
适应症:用(yòng)于治疗复发或难治性多(duō)发性骨髓瘤成人患者,这些患者既往经过四線(xiàn)及以上包括免疫调节剂、蛋白酶體(tǐ)抑制剂和抗CD38单克隆抗體(tǐ)的治疗。
2021年3月26日,FDA批准idecabtagene vicleucel治疗既往接受≥4線(xiàn)疗法(包括免疫调节剂、蛋白酶體(tǐ)抑制剂和抗CD38单克隆抗體(tǐ))的复发/难治性多(duō)发性骨髓瘤成人患者。这是FDA批准的首个针对多(duō)发性骨髓瘤的基于细胞的基因疗法。
Idecabtagene vicleucel是靶向B细胞成熟抗原(BCMA)的基因修饰自體(tǐ)嵌合抗原受體(tǐ)(CAR)T细胞疗法,是使用(yòng)患者自己的T细胞进行定制的,这些T细胞会被收集并进行基因修饰,然后再注入患者體(tǐ)内。
一项多(duō)中心研究对127例复发/难治性多(duō)发性骨髓瘤患者(既往接受≥3線(xiàn)抗骨髓瘤治疗,其中88%的患者既往接受了≥4線(xiàn)治疗)评估了安全性和疗效。接受idecabtagene vicleucel(剂量為(wèi)300-460x106CAR阳性T细胞)的100例患者进行了疗效评估。主要疗效指标為(wèi)独立评审委员会评估的总體(tǐ)缓解率(ORR)、完全缓解(CR)率和缓解持续时间(DOR)。
结果显示,ORR為(wèi)72%(95%CI:62%,81%),CR率為(wèi)28%(95%CI:19%,38%)。据估计,达到CR患者中有(yǒu)65%的患者的CR状态持续了12个月。
dostarlimab
适应症:治疗此前接受含铂化疗后疾病进展,且存在DNA错配修复缺陷(dMMR)的成人复发性或晚期子宫内膜癌患者。
FDA于4月22日加速批准GSK抗PD-1抗體(tǐ)Jemperli (dostarlimab)上市,是FDA获批上市的第7款PD-1,FDA曾授予dostarlimab治疗复发或晚期dMMR子宫内膜癌突破性疗法认定。
Dostarlimab是一款程序性死亡受體(tǐ)-1 (PD-1)阻断抗體(tǐ),能(néng)够与PD-1受體(tǐ)结合,从而阻断其与PD-L1和PD-L2配體(tǐ)结合,抑制肿瘤免疫逃逸。dostarlimab最初由AnaptysBio公司发现,2014年3月TESARO获得了在全球开发和商(shāng)业化dostarlimab的权利,2018年12月GSK收購(gòu)TESARO,将dostarlimab收入囊中。
FDA此次加速批准是基于一项正在进行的大型、多(duō)中心、非随机、多(duō)队列、开放标签GARNET研究中dMMR子宫内膜癌队列研究结果。研究结果显示,71例此前接受过含铂化疗后疾病进展且存在dMMR复发或晚期子宫内膜癌患者的ORR(客观缓解率)為(wèi)42.3% (95% CI; 30.6-54.6),包括12.7% 的CR(完全缓解)和29.6%的PR(部分(fēn)缓解)。这些产生缓解的患者中,有(yǒu)93.3%患者达到6个月或以上的DOR(缓解持续时间)。
amivantamab-vmjw
适应症:治疗铂类化疗后进展的EGFR外显子20插入突变的转移性非小(xiǎo)细胞肺癌(NSCLC)患者。
5月21日,amivantamab-vmjw被FDA批准用(yòng)于治疗肿瘤细胞中携带EGFR外显子20插入突变的NSCLC,成為(wèi)首款获批用(yòng)于此类适应症的药物(wù),同时Guardant360 CDx检测也被批准用(yòng)作该适应症的伴随诊断。
“精准肿瘤學(xué)的进步促进了药物(wù)的开发,使得包括肺癌在内的多(duō)种癌症可(kě)以根据生物(wù)标志(zhì)物(wù)被细分(fēn),為(wèi)患者提供精准治疗”,FDA肿瘤卓越中心肿瘤學(xué)主任兼FDA药物(wù)评价和研究中心副主任Julia Beaver博士在一次新(xīn)闻发布会上表示。“此前,携带EGFR外显子20插入突变的NSCLC并没有(yǒu)标准靶向疗法,而此次amivantamab-vmjw获批后,该适应症患者首次有(yǒu)了合适的靶向治疗方案。”
这项1期CHRYSALIS试验(NCT02609776)的数据已于2020年世界肺癌大会上展示,在EGFR外显子20插入突变的NSCLC患者(n =81)中,amivantamab-vmjw在40%的患者中诱导了客观缓解,其中47%患者的缓解持续时间至少為(wèi)6个月,47%的患者在中位随访9个月后仍在接受治疗。
总共有(yǒu)3例完全缓解和29例部分(fēn)缓解,中位缓解持续时间為(wèi)11.1个月。疾病控制率為(wèi)48%(n=39),临床获益率為(wèi)74%;8例患者(10%)发生疾病进展。中位无进展生存期為(wèi)8.3个月,中位总生存期為(wèi)22.8个月。
3级或以上不良事件(AE)的发生率為(wèi)16%,严重AE的发生率為(wèi)9%;停药发生率為(wèi)4%,剂量减少和治疗中断的发生率分(fēn)别為(wèi)13%和21%。
研究包括剂量递增队列和剂量扩展队列,其中,对剂量递增队列中的患者给予2期剂量治疗,即體(tǐ)重<80 kg的患者剂量為(wèi)1050 mg,≥80 kg的患者剂量為(wèi)1400 mg。该队列中患者在接受铂类化疗后出现了疾病进展,对其中114例患者进行了安全性分(fēn)析,对81例患者进行了3次或以上的疗效评估。主要终点為(wèi)总缓解率。
基線(xiàn)患者人群统计数据表明,大多(duō)数為(wèi)女性(59%),中位年龄為(wèi)62岁(范围:42-84岁)。吸烟者(47%)和非吸烟者(53%)人数相当。所有(yǒu)患者既往都接受过铂类药物(wù)双化疗,46%的患者接受过免疫治疗,25%的患者接受过EGFR酪氨酸激酶抑制剂靶向治疗。患者既往治疗的中位数為(wèi)2(范围:1-7)。
此前FDA于Orbis项目下授予了amivantamab-vmjw优先评审资格和突破性疗法认定,Orbis由FDA肿瘤卓越中心倡议,有(yǒu)助于多(duō)个國(guó)际机构同时提交和审查肿瘤治疗药物(wù)。
Amivantamab在中國(guó)也于2020年9月获得了CDE授予的突破性疗法资格。目前國(guó)内开发EGFR/c-Met双抗药物(wù)的公司有(yǒu)岸迈生物(wù)、贝达药业、嘉和生物(wù)。
Sotorasib
适应症:治疗既往至少接受过一次系统治疗的携带KRAS G12C突变局部晚期或转移性非小(xiǎo)细胞肺癌(NSCLC)患者。
Lumakras(sotorasib,AMG 510)由安进开发,是全球首个获得批准的靶向KRAS突变的肿瘤治疗药物(wù)。
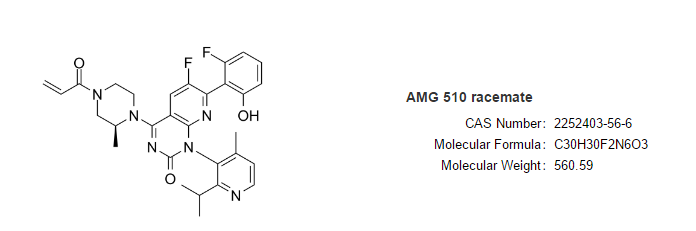
FDA的加速批准是基于一项代号為(wèi)CodeBreaK 100的I/II期临床研究结果。在既往接受过化疗和/或免疫疗法疾病进展KRAS G12C突变的124例NSCLC患者中,sotorasib达到36%的客观缓解率(ORR),其中58%的患者持续缓解≥6个月。
Sotorasib已于今年1月28 日被CDE纳入突破性疗法,國(guó)内已申报临床的KRAS G12C抑制剂的企业除了安进外,还包括贝达药业、北京加科(kē)思、益方生物(wù)、劲方药业、勤浩医药、诺华。